Introdução
As dermatoses purpúricas pigmentadas (PPD) são um grupo raro de doenças crónicas e benignas caracterizadas por múltiplas petéquias em máculas hiperpigmentadas, amarelo-acastanhadas.1 As diferentes variantes são formas clínicas distintas da mesma entidade com características histopatológicas semelhantes.2,3
Existem 5 variantes clássicas: Doença de Schamberg (PPD progressiva), púrpura eczematóide de Doucas e Kapetanakis (púrpura pruriginosa), dermatose liquenóide pigmentada de Gougerot e Blum, líquen aureus (líquen purpúrico), e doença de Majocchi (púrpura anularis telangiectodes).1 As variantes menos comuns são PPD granulomatosa, púrpura com prurido de Loewenthal, PPD linear, PPD transitória, e PPD familiar.2
Epidemiologia
PPDs são raras e afectam predominantemente adultos,1 embora tenham sido notificados casos em crianças.4 A variante mais comum tanto em adultos como em crianças é a doença de Schamberg. As PPD, e as formas lineares em particular,6 são geralmente mais comuns nos homens.1 A doença de Majocchi é mais comum nas mulheres.
Etiologia e Patogénese
Embora as causas das PPD sejam desconhecidas, foi proposta uma série de factores desencadeantes, incluindo exercício, hipertensão venosa, diabetes mellitus, infecções,2,7 e vários medicamentos (Quadro 1).8 Foi notificada uma associação com dislipidemia e doenças auto-imunes para PPD granulomatosas.9 Na maioria dos casos, contudo, não foi identificada qualquer causa.10
| Sedativos | Fenobarbital, clordiazepóxido, meprobamato |
| Tiamina (B1) | |
| Furosemida | |
| Nitroglicerina, bezafibrato, hidralazina, dipiridamol, sildenafil | |
| Antibióticos | Ampicilina | Analgésicos | Anti-inflamatórios não-esteróides, aspirina, acetaminofenol |
| Estimulantes | Pseudoefedrina |
| Hormonas | Medroxiprogesterona acetato |
| Antidiabéticos | Glipizide |
| Agentes de quimioterapia | Topical 5-fluorouracil |
| Antivirals | Interferon α |
| Retinoids | Isotretinoin |
Source: Kaplan et al.8
Dilatação capilar e fragilidade foram atribuídas um possível papel patogénico nos PPD.10 Foi levantada a hipótese de que as células responsáveis por estas perturbações são células envolvidas na estrutura dos vasos sanguíneos, tais como fibroblastos e células endoteliais. Quer por activação (por exemplo, pressão intravascular elevada) quer espontaneamente, a função destas células pode ser alterada, causando a fuga de glóbulos vermelhos (hemácias) através das paredes dos vasos,11 desencadeando uma reacção de hipersensibilidade mediada por células. A resposta imunitária mediada por células parece, portanto, ter um papel fundamental na patogénese das PPD.12,13 O infiltrado inflamatório perivascular é constituído por células CD4+ T14 (com expressão CD7 reduzida15) e células CD1a+ dendríticas.12
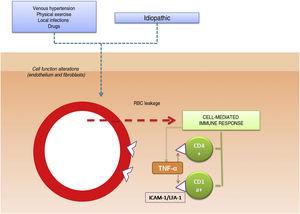
O papel patogénico das moléculas de adesão celular nas PPD também tem sido analisado em vários estudos. As CAM são proteínas de membrana que interagem com ligandos específicos que fornecem e mantêm contacto entre diferentes células e entre células e proteínas de matriz extracelular. Foram observados altos níveis de expressão para moléculas de adesão LFA-1 (antigénio-1 da função linfocitária) e ICAM-1 (molécula de adesão intercelular-1) em células inflamatórias e para ICAM-1 e ELAM-1 (molécula de adesão leucocitária endotelial- 1)12 em células endoteliais. As células T activadas por um estímulo antigénico adeririam assim às células endoteliais, fibroblastos e queratinócitos.16 As citocinas produzidas por leucócitos (por exemplo, o factor de necrose tumoral α) podem desencadear a expressão destas moléculas de adesão (Fig. 1).
 br>>>div>
br>>>div>
Mecanismo etiológico e patogénico. Uma das hipóteses mais amplamente aceites é que as células T são activadas por um estímulo antigénico e ligam-se às células endoteliais, fibroblastos, e queratinócitos através da expressão de moléculas de adesão. TNF-α indica factor de necrose tumoral α ; ICAM-1, molécula de adesão intercelular-1; LFA-1, antigénio de função linfocitária-1.
As citocinas acima referidas podem também resultar numa diminuição da libertação do activador do plasminogénio endotelial e/ou num aumento excessivo do inibidor do activador do plasminogénio,17 levando à redução da actividade fibrinolítica e da deposição intraperivascular de fibrina observada nas PPD.18
A imunofluorescência directa pode mostrar fibrinogénio, imunoglobulina M, e/ou deposição C3 nos vasos dérmicos superficiais.10
Uma outra hipótese que surgiu nos últimos anos é que as PPD podem representar uma alteração epiteliotropica insidiosa das células T. Esta teoria é apoiada pela observação do epidermotropismo ou de um padrão monoclonal no infiltrado inflamatório.15,19 Houve mesmo alguns relatos de progressão para micose fungoides.20-22 Como é difícil distinguir entre micose fungoides purpúricas e PPD monoclonais, é essencial integrar os achados clínicos, moleculares e histopatológicos.15,20,23 Poikiloderma, prurido, placas coalescentes, uma duração superior a 1 ano, um padrão monoclonal, e diminuição da expressão CD7 e CD62 L no infiltrado devem todos levantar suspeitas de progressão da doença, mesmo na ausência de atipia linfocítica explícita.15,24 Alguns autores optam por tratar DPP disseminado e monoclonal como micose fungoides em fase inicial.
Variantes clínicasProgressivas PPD ou doença de Schamberg1,2
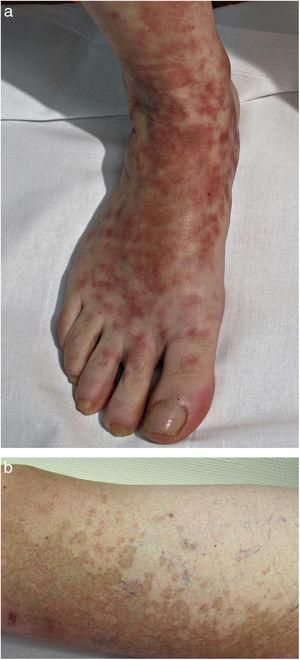
Em PPD progressivo ou doença de Schamberg, as lesões aparecem geralmente em ambas as extremidades inferiores, mas também podem afectar o tronco, braços, coxas, ou nádegas. Apresentam-se como máculas vermelho-alaranjadas com manchas periféricas purpúreas que se assemelham a grãos de pimenta-de-caiena (Fig. 2A e B); estas manchas adquirem uma cor castanha-amarelada à medida que progridem. As lesões são geralmente assintomáticas, embora alguns pacientes descrevam o prurido. Seguem um curso crónico com numerosas recaídas e remissões.
>>div>
doença de Schamberg. A, Máculas vermelho-laranja que tendem a coalescer, formando grandes placas. B, Manchas periféricas de pimenta-de-caiena.
Púrpura Pruriginosa ou Púrpura Eczematóide de Doucas e Kapetanakis1,25
Púrpura Pruriginosa ou Púrpura Eczematóide de Doucas e Kapetanakis é a variante mais extensa e pruriginosa dos PPD. Afecta principalmente as extremidades inferiores e é clinicamente semelhante à doença de Schamberg, com máculas purpúricas ou petequiais mas com uma superfície de escamação. A púrpura eczematóide de Doucas e Kapetanakis tem sido associada à dermatite de contacto alérgica à borracha e ao vestuário. Onset é rápido (15-30 dias) e as lesões podem durar meses ou anos.
Dermatose liquenóide pigmentada de Gougerot e Blum1,2,26
Dermatose liquenóide pigmentada de Gougerot e Blum é caracterizada por pápulas liquenóides violáceas que tendem a fundir-se, formando grandes placas que estão normalmente localizadas nas pernas, mas que podem afectar o tronco. A condição segue um curso crónico e normalmente afecta os homens idosos. Deve ser distinguido de Kaposi sarcoma.
Lichen Aureus ou Lichen Purpuricus1,2,27
Lichen aureus ou Lichen purpuricus é uma variante mais localizada do PPD. As lesões são persistentes e são tipicamente solitárias ou pequenas em número. O líquen aureus é caracterizado pelo aparecimento súbito de pequenas pápulas amarelo-orangoladas com um aspecto liquenóide e uma tendência a coalescer em placas com dimensões entre 1 e 20 cm associadas a lesões purpúricas milimétricas (Fig. 3). A condição afecta principalmente as extremidades inferiores, mas as lesões podem ocorrer em qualquer parte do corpo. São geralmente assintomáticas. As variantes zosteriformes28 e segmentares ao longo das linhas de Blaschko29 ou seguindo o curso da safena30 ou veias cefálicas31 foram descritas em crianças e adolescentes.
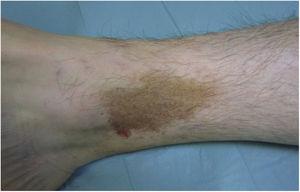
Lichen aureus. Placa solitária constituída por pequenas máculas amarelo-laranja coalescentes na perna.
Purpura Annularis Telangiectodes ou doença de Majocchi1,2,25
Purpura annularis telangiectodes ou doença de Majocchi apresenta-se com máculas anulares vermelhas-violáceas (Fig. 4), seguidas de puncta telangiectásica vermelha mais escura. As lesões estendem-se de forma periférica e o seu centro desvanece-se gradualmente, podendo adquirir um aspecto atrófico. A erupção começa nas extremidades inferiores e depois espalha-se para o tronco e braços; caracteriza-se por um grande número de lesões. Foi descrita uma variante conhecida como arciform purpura annularis telangiectodes com menos mas maiores lesões, com uma morfologia arqueada característica.32
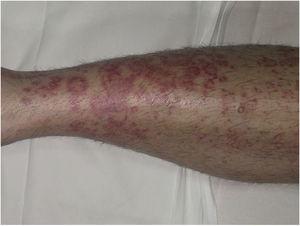
Doença de Majocchi. Lesões vermelhas-violáceas anulares na perna.
Outras Variantes
Hersch e Schwayder33 descreveram o que é considerado como uma forma linear, unilateral e rara, que deve ser diferenciada das formas lineares da doença de Schamberg e do líquen aureus. Higgins e Cox34 descreveram uma forma quadrantica que atribuíram a uma obstrução vascular na pélvis.
Têm também havido relatos de uma variante transitória35 , incluindo entidades como o angioma serpiginosum,36 que é uma desordem vascular pouco comum que normalmente começa na infância, é mais comum nas mulheres, e mostra evidência de dependência do estrogénio. O serpiginoso angioma é caracterizado por múltiplas máculas vermelhas-púrpuras assintomáticas dispostas em pequenos grupos seguindo um padrão de serpiginoso ao longo das extremidades.
Púrpura de Loewenthal,37 que só foi descrita em adultos, é considerada uma variante mais sintomática da doença de Schamberg.
DPPranulomatosa, descrita por Saito,38 é uma forma histopatológica mais comum nas mulheres e clinicamente indistinguível de outras DPPs.
Finalmente, tem havido relatos de formas familiares autossómicas dominantes da doença de Schamberg e purpura annularis telangiectoides.39
Histopatologia
Histopatologicamente, as PPDs caracterizam-se por um infiltrado linfocitário perivascular centrado nos pequenos vasos superficiais. Outros achados típicos são inchaço endotelial, estreitamento luminal,10 hemácias extravasadas, e macrófagos carregados de hemossiderina (Fig. 5 A e B). A coloração de Perls e Fontana-Masson mostra depósitos de hemossiderina (ferro) na derme superficial, distinguindo a DPP da dermatite de estase, que tem depósitos mais profundos.2
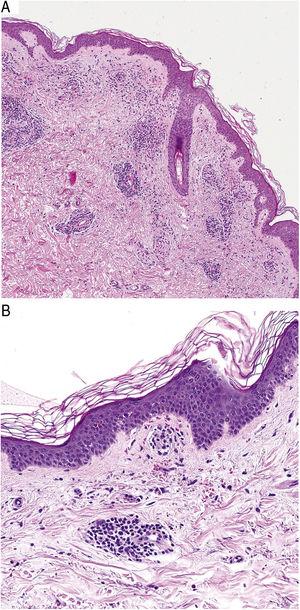
>div>
Características histopatológicas da doença de Schamberg. A, Infiltrado envolvendo pequenos vasos na derme superficial. B, Infiltrado linfocitário, com estreitamento luminal e eritrócitos extravasados.
Um achado característico de líquen aureus é uma epiderme intacta separada de um infiltrado dérmico semelhante a uma banda por uma área de tecido conjuntivo poupado (área de Grenz)27 (Fig. 6). Este infiltrado é também típico na dermatose liquenóide pigmentada de Gougerot e Blum.26 A púrpura eczematóide de Doucas e Kapetanakis, pelo contrário, apresenta esponjoses epidérmicas e neutrófilos no infiltrado.25 O PPD granulomatoso caracteriza-se por um infiltrado granulomatoso perivascular sobre características típicas.38 Uma comparação de características clínicas, localização e achados histopatológicos para as variantes mais comuns de PPD é fornecida na Tabela 2.
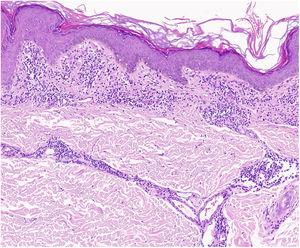
Características histopatológicas do líquen aureus. Infiltrado tipo banda na derme papilar e infiltrado perivascular superficial.
| Variante | Apresentação Clínica | Localização | Resultados histopatológicos |
|---|---|---|---|
| Doença de Schamberg | Red-máculas cor de laranja com manchas periféricas que se assemelham a grãos de pimenta de caiena | Membros inferiores e ocasionalmente tronco, braços, coxas e nádegas | |
| Eczematoid purpura de Doucas e Kapetanakis | Similar às manifestações da doença de Schamberg mas com escamação e prurido intenso | Baixos membros | Infiltrair com mais número de neutrófilos e esponjas epidérmicas |
| Dermatose liquenóide pigmentada de Gougerot e Blum | Pápulas liquenóides voláceas que se fundem para formar placas | Membros inferiores | Bandcomo infiltrado dérmico |
| Lichen aureus | Iolado persistente vermelho-placa laranja e lesões purpúricas | Lembros inferiores | Epidermes e bandas não trocadas…como infiltrado dérmico com área de Grenz |
| Doença de Majocchi | Placas perifericamente extensíveis com punta telangiectásica nas extremidades e desvanecimento na área central | Membros inferiores e tronco | Idêntico à doença de Schamberg |
Enquanto alguns autores1,13 considera a capilaridade como uma característica determinante dos DPPs, Ackerman40 não acredita que este seja o caso, uma vez que existe uma ausência de fibrina na parede luminal e trombos na lumina.
Diagnóstico
Além da biopsia da pele, recomenda-se um exame de sangue para excluir trombocitopenia, coagulação ou doenças auto-imunes (anticorpos antinucleares, factor reumatóide), e infecções crónicas (anti-HCV e anti-HBsAg).2
Diagnóstico diferencial
O diagnóstico diferencial deve incluir outras condições que apresentem manifestações purpúricas envolvendo as extremidades inferiores. Estas entidades e as suas principais características estão resumidas na Tabela 3.
| Entidades Científicas | Características principais |
|---|---|
| Drug-reacções de hipersensibilidade | Uso recente de droga causal |
| Carbamazepina, meprobamato, clordiazepóxido, furosemida, nitroglicerina, vitamina B1, e 5-fluorouracil tópico | |
| Dermatite de contacto purpúrica ao vestuário | Lesões confinadas a áreas da pele em contacto com o vestuário; prurido intenso |
| Sinais de insuficiência venosa crónica: inchaço, varizes, sensação de peso, úlceras venosas | |
| Associada com contagem de plaquetas | |
| Em pacientes idosos, A púrpura pode ser associada a antiplaquetário, anticoagulante, ou uso de corticosteróides | |
| Exantema purpúrico devido a infecção viral | Outros sinais de infecção |
| Vasculite leucocitocíclica | Lesões purpúricas palpáveis |
| Púrpura de Schönlein-Henoch | Idade, 3-15 anos; púrpura simétrica que afecta pernas e nádegas; dores nas articulações e abdominais |
| Afecta doentes idosos ou imunodeprimidos | |
| Micose fungóide purpúrica | Onset > 1 ano; divulgado, perfil monoclonal em infiltração e perda de CD7 |
Source: Sardana et al.2, Kim et al.,25 e Risikesan et al.26
Dermoscopia
p>O achado dermoscópico mais comum é um fundo cobre-vermelho difuso que corresponde histopatologicamente ao infiltrado dérmico linfocitário, hemácias extravasadas, e macrófagos carregados de hemossiderina.41 Outras descobertas incluem glóbulos vermelhos e pontos, que podem ser explicados pelas hemácias extravasadas, o aumento do número de vasos sanguíneos, e a dilatação destes vasos42 (Fig. 7). Os pontos castanhos são observados em quase 50% dos doentes e correspondem à disposição esférica ou elíptica dos melanócitos e melanófagos na junção dermoepidérmica. Num terço dos casos, a dermopediatria mostra uma pseudo-rede pigmentada que corresponde à camada de células basais hiperpigmentadas e incontinência pigmentar na derme papilar. Os resultados dermoscópicos específicos relatados para o líquen aureus incluem um fundo vermelho acobreado com pontos e glóbulos castanhos e vermelhos, pontos cinzentos, e uma pseudo-rede compreendendo linhas pigmentadas interconectadas43 (Fig. 8).
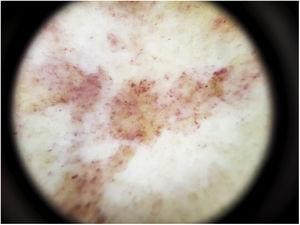
Características dermoscópicas da doença de Schamberg. Fundo vermelho-cobre e glóbulos vermelhos.
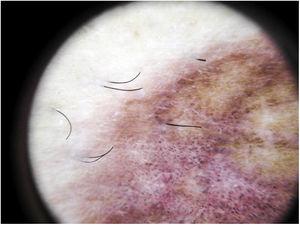
Características termoscópicas do líquen aureus. Fundo vermelho-acastanhado com pontos cinzentos e uma pseudo-rede pigmentada.
Tratamento
considerando que os DPPs são benignos e que não existem tratamentos padronizados com eficácia comprovada, os riscos e benefícios de qualquer tratamento devem ser cuidadosamente ponderados.
Dev>A natureza benigna e largamente assintomática dos DPPs, nenhum tratamento é uma opção.4 O tratamento, contudo, é frequentemente necessário devido à natureza crónica da doença, aos seus impactos físicos e psicológicos, e à presença de lesões extensas ou prurido.
A maioria das recomendações de tratamento baseiam-se em pequenas séries de casos, e nenhuma delas é apoiada por provas suficientes para ser considerada um tratamento universal.
Uma gama de tratamentos tópicos e sistémicos, detalhados abaixo, foram descritos em pequenas séries e relatórios de casos.
Tratamentos tópicosCorticóides tópicos
Corticóides tópicos são os tratamentos mais comuns descritos e têm sido observados para reduzir a comichão e, em alguns casos, lesões claras.2,4
Os agentes mais utilizados são corticosteróides de média e alta potência (clobetasol e aceponato de metilprednisolona)
Inibidores de calcineurina tópicos
Aplicação tópica de tacrolimus44 e pimecrolimus45 durante vários meses foi encontrada para resolver o líquen aureus.
Dada a natureza crónica das lesões PPD e a necessidade de tratamento a longo prazo, os inibidores de calcineurina podem ser considerados uma boa alternativa aos corticosteróides tópicos para limpar ou resolver lesões.
Fototerapia
Fototerapia é uma boa opção para tratar doenças extensivas ou PPD que não respondem aos corticosteróides tópicos ou inibidores de calcineurina.
P>Posicionou-se que a fototerapia pode ser eficaz porque produz um efeito imunomodulador que modifica a actividade das células T e reduz a produção de interleucina-2, resultando em melhoria.46
O tratamento Psoralen e UV-A (PUVA) tem sido utilizado com sucesso em doentes com doença de Schamberg, dermatose liquenóide purpúrica, e líquen aureus. Na série publicada até à data, entre 7 e 29 sessões com doses cumulativas que variam entre 16 e 49 J/cm2 foram necessárias para se conseguir a remissão. O novo tratamento também se revelou eficaz, e em alguns casos foi necessário um tratamento de manutenção durante vários meses para prolongar a resposta.2,47-49
Fototerapia UV-B de banda larga com doses cumulativas entre 11 e 49 J/cm2 administradas em 24 a 60 sessões produziu respostas favoráveis em pacientes com diferentes variantes clínicas de DPP. Tal como no tratamento PUVA, houve relatos de recorrência após a interrupção do tratamento mas uma boa resposta ao novo tratamento.46,50,51
Terapia UV-B de banda estreita é considerada uma boa opção devido aos seus poucos efeitos adversos e ao seu bom perfil de tolerabilidade. Por conseguinte, deve ser considerada como uma opção para pacientes pediátricos, pacientes com lesões extensas, e pacientes resistentes a tratamentos tópicos.5,52
Tratamentos sistémicosPentoxifilina
Há relatos de PPD em resposta à pentoxifilina oral. Tem sido sugerido que a pentoxifilina pode ser eficaz porque inibe a adesão das células T ao endotélio vascular por interacção com ICAM-1,53,54
Pentoxifilina tem sido utilizada sozinha, numa dose de 400 mg duas ou três vezes ao dia durante 2 a 3 meses,42,43 ou em combinação com outros medicamentos, tais como prostaciclinas (prostaglandina I1)55 e corticosteróides orais.56 A pentoxifilina também foi considerada ineficaz no tratamento de DPPs.57
Ácido ascórbico e bioflavonóides (Rutina/Rutoside)
Asso ácido ascórbico e bioflavonóides aumentam a produção de colagénio, reduzindo assim a permeabilidade vascular e melhorando a função da barreira endotelial vascular, doses elevadas de vitamina C combinadas com um glicosídeo flavonóide (como a rutoside/rutina), presente nos citrinos, administrado ao longo de vários meses, resultaram em melhorias clínicas e, em alguns casos, na resolução.58
Outros Tratamentos
Têm havido relatos isolados de resposta a vários tratamentos sistémicos, tais como griseofulvin,59 colchicina,60 metotrexato,61 e ciclosporina.62
Conclusões
Os DPPs são uma condição dermatológica comum e têm um grande impacto na qualidade de vida dos pacientes, devido tanto aos sintomas como às preocupações cosméticas. Embora as diferentes variantes sejam clinicamente muito semelhantes, existem várias características clínicas, histopatológicas e dermoscópicas que ajudam a estabelecer um diagnóstico mais específico.
Finalmente, embora não haja provas suficientes na literatura para recomendar qualquer tratamento como tratamento de primeira linha, existem numerosas opções que podem alcançar melhorias consideráveis.
Conflitos de interesse
Os autores declaram não ter conflitos de interesse.