Introduction
Les dermatoses purpuriques pigmentées (PPD) sont un groupe rare de maladies chroniques et bénignes caractérisées par de multiples pétéchies sur des macules hyperpigmentées de couleur jaune-brun.1 Les différentes variantes sont des formes cliniques distinctes de la même entité avec des caractéristiques histopathologiques similaires.2,3
Il existe 5 variantes classiques : La maladie de Schamberg (PPD progressive), le purpura eczématoïde de Doucas et Kapetanakis (purpura prurigineux), la dermatose lichénoïde purpurique pigmentée de Gougerot et Blum, le lichen aureus (lichen purpuricus) et la maladie de Majocchi (purpura annularis telangiectodes).Les variantes moins fréquentes sont la PPD granulomateuse, le purpura prurigineux de Loewenthal, la PPD linéaire, la PPD transitoire et la PPD familiale2.
Epidémiologie
Les PPD sont rares et touchent principalement les adultes,1 bien que des cas aient été rapportés chez les enfants.4 La variante la plus fréquente chez les adultes et les enfants est la maladie de Schamberg. Les PPD, et les formes linéaires en particulier6, sont généralement plus fréquentes chez les hommes1. La maladie de Majocchi est plus fréquente chez les femmes.
Etiologie et pathogénie
Bien que les causes des PPD soient inconnues, un éventail de facteurs déclenchants a été proposé, notamment l’exercice, l’hypertension veineuse, le diabète sucré, les infections,2,7 et divers médicaments (tableau 1).8 Une association avec la dyslipidémie et les maladies auto-immunes a été rapportée pour les PPD granulomateuses.9 Dans la plupart des cas, cependant, aucune cause n’est identifiée10.
Médicaments associés aux dermatoses purpuriques pigmentées.
| Sédatifs | Phénobarbital, chlordiazépoxide, méprobamate | Vitamines | Thiamine (B1) | Diurétiques | Furosémide |
| Médicaments cardiovasculaires | Nitroglycérine, bézafibrate, hydralazine, dipyridamole, sildénafil | Antibiotiques | Ampicilline | Analgésiques | Anti-inflammatoires non stéroïdiens, l’aspirine, acétaminophénol | Stimulants | Pseudoéphédrine |
| Hormones | Medroxyprogestérone acétate | Médicaments antidiabétiques | Glipizide | Agents de chimiothérapie | Topique 5-fluorouracile | Antiviraux | Interféron α | Rétinoïdes | Isotrétinoïne |
Source : Kaplan et al.8
La dilatation et la fragilité des capillaires se sont vues attribuer un rôle pathogène possible dans les DPP.10 On a émis l’hypothèse que les cellules responsables de ces troubles sont des cellules impliquées dans la structure des vaisseaux sanguins, comme les fibroblastes et les cellules endothéliales. Que ce soit par activation (par exemple, en cas de pression intravasculaire élevée) ou spontanément, la fonction de ces cellules peut être altérée, entraînant une fuite des globules rouges (GR) à travers les parois des vaisseaux,11 déclenchant une réaction d’hypersensibilité à médiation cellulaire. La réponse immunitaire à médiation cellulaire semblerait donc avoir un rôle fondamental dans la pathogenèse des DPP.12,13 L’infiltrat inflammatoire périvasculaire est composé de lymphocytes T CD4+14 (avec une expression réduite de CD715) et de cellules dendritiques CD1a+.12
Le rôle pathogène des molécules d’adhésion cellulaire dans les DPP a également été analysé dans plusieurs études. Les CAM sont des protéines membranaires qui interagissent avec des ligands spécifiques qui assurent et maintiennent le contact entre différentes cellules et entre les cellules et les protéines de la matrice extracellulaire. Des niveaux d’expression élevés ont été observés pour les molécules d’adhésion LFA-1 (lymphocyte function antigen-1) et ICAM-1 (intercellular adhesion molecule-1) dans les cellules inflammatoires et pour ICAM-1 et ELAM-1 (endothelial leukocyte adhesion molecule- 1)12 dans les cellules endothéliales. Les cellules T activées par un stimulus antigénique adhéreraient ainsi aux cellules endothéliales, aux fibroblastes et aux kératinocytes.16 Les cytokines produites par les leucocytes (par exemple, le facteur α de nécrose tumorale) peuvent déclencher l’expression de ces molécules d’adhésion (figure 1).
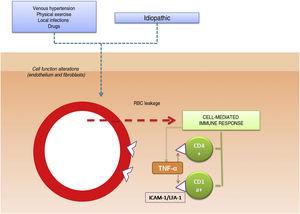
Mécanisme étiologique et pathogénique. L’une des hypothèses les plus largement acceptées est que les cellules T sont activées par un stimulus antigénique et se lient aux cellules endothéliales, aux fibroblastes et aux kératinocytes par l’expression de molécules d’adhésion. TNF-α indique le facteur de nécrose tumorale α ; ICAM-1, molécule d’adhésion intercellulaire-1 ; LFA-1, antigène de fonction lymphocytaire-1.
Les cytokines susmentionnées peuvent également entraîner une diminution de la libération de l’activateur du plasminogène endothélial et/ou une augmentation excessive de l’inhibiteur de l’activateur du plasminogène17, ce qui conduit à la réduction de l’activité fibrinolytique et au dépôt intra-périvasculaire de fibrine observé dans les PPD18.
L’immunofluorescence directe peut montrer un dépôt de fibrinogène, d’immunoglobuline M et/ou de C3 dans les vaisseaux dermiques superficiels.10
Une autre hypothèse qui a émergé ces dernières années est que les PPD pourraient représenter une altération insidieuse des cellules T épithéliotropes. Cette théorie est soutenue par l’observation d’un épidermotropisme ou d’un motif monoclonal dans l’infiltrat inflammatoire.15,19 On a même rapporté des cas d’évolution vers un mycosis fongoïde.20-22 Comme il est difficile de faire la distinction entre un mycosis fongoïde purpurique et une PPD monoclonale, il est essentiel d’intégrer les résultats cliniques, moléculaires et histopathologiques.15,20,23 Une poïkilodermie, un prurit, des plaques coalescentes, une durée de plus d’un an, un modèle monoclonal et une diminution de l’expression des CD7 et CD62 L dans l’infiltrat doivent faire suspecter une progression de la maladie, même en l’absence d’atypies lymphocytaires manifestes15,24. Certains auteurs choisissent de traiter la DPP disséminée et monoclonale comme un mycosis fongoïde de stade précoce.
Variantes cliniquesDPP progressive ou maladie de Schamberg1,2
Dans la DPP progressive ou maladie de Schamberg, les lésions apparaissent généralement sur les deux extrémités inférieures, mais elles peuvent également affecter le tronc, les bras, les cuisses ou les fesses. Elles se présentent sous la forme de macules rouge-orange avec des taches purpuriques périphériques ressemblant à des grains de poivre de Cayenne (Fig. 2A et B) ; ces taches acquièrent une couleur brun-jaune au fur et à mesure de leur progression. Les lésions sont généralement asymptomatiques, bien que certains patients décrivent un prurit. Elles suivent une évolution chronique avec de nombreuses rechutes et rémissions.
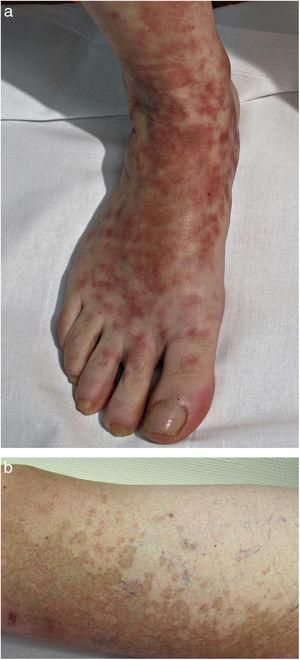
Maladie de Schamberg. A, macules rouge-orange qui ont tendance à coalescer, formant de grandes plaques. B, taches périphériques au poivre de Cayenne.
Purpura prurigineux ou purpura eczématoïde de Doucas et Kapetanakis1,25
Le purpura prurigineux ou purpura eczématoïde de Doucas et Kapetanakis est la variante la plus étendue et la plus prurigineuse des PPD. Il affecte principalement les extrémités inférieures et est cliniquement similaire à la maladie de Schamberg, avec des macules purpuriques ou pétéchiales mais avec une surface écaillée. Le purpura eczématoïde de Doucas et Kapetanakis a été associé à une dermatite de contact allergique au caoutchouc et aux vêtements. L’apparition est rapide (15 à 30 jours) et les lésions peuvent durer des mois ou des années.
Dermatose lichénoïde purpurique pigmentée de Gougerot et Blum1,2,26
La dermatose lichénoïde purpurique pigmentée de Gougerot et Blum est caractérisée par des papules lichénoïdes violacées qui ont tendance à fusionner, formant de grandes plaques qui sont généralement situées sur les jambes mais peuvent affecter le tronc. L’affection suit une évolution chronique et touche généralement les hommes âgés. Elle doit être distinguée du sarcome de Kaposi.
Lichen aureus ou lichen purpurique1,2,27
Le lichen aureus ou lichen purpurique est une variante plus localisée de la PPD. Les lésions sont persistantes et sont typiquement solitaires ou en petit nombre. Le lichen aureus se caractérise par l’apparition soudaine de petites papules jaunes-orangées d’aspect lichénoïde et ayant tendance à coalescer en plaques mesurant entre 1 et 20 cm associées à des lésions purpuriques millimétriques (Fig. 3). L’affection touche principalement les extrémités inférieures, mais les lésions peuvent survenir sur n’importe quelle partie du corps. Elles sont généralement asymptomatiques. Des variantes zostériformes28 et segmentaires selon les lignes de Blaschko29 ou suivant le trajet des veines saphènes30 ou céphaliques31 ont été décrites chez les enfants et les adolescents.
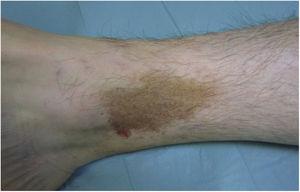
Lichen aureus. Plaque solitaire comprenant de petites macules jaune-orange coalescentes sur la jambe.
Purpura annularis telangiectodes ou maladie de Majocchi1,2,25
Purpura annularis telangiectodes ou maladie de Majocchi se présente sous la forme de macules annulaires rouge-violacé (Fig. 4), suivies de ponctuations télangiectasiques rouge plus foncé. Les lésions s’étendent en périphérie et leur centre s’estompe progressivement et peut acquérir un aspect atrophique. L’éruption commence dans les extrémités inférieures et s’étend ensuite au tronc et aux bras ; elle se caractérise par un grand nombre de lésions. Une variante connue sous le nom de purpura annularis telangiectodes arciforme présentant des lésions moins nombreuses mais plus grandes avec une morphologie arquée caractéristique a été décrite.32
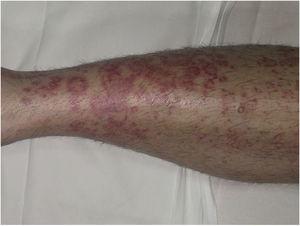
Maladie de Majocchi. Lésions annulaires rouge-violacées sur la jambe.
Autres variantes
Hersch et Schwayder33 ont décrit ce qui est considéré comme une forme linéaire rare, unilatérale, qui doit être différenciée des formes linéaires de la maladie de Schamberg et du lichen aureus. Higgins et Cox34 ont décrit une forme quadratique qu’ils ont attribuée à une obstruction vasculaire dans le bassin.
On a également signalé une variante transitoire35, y compris des entités telles que l’angiome serpiginosum36, qui est un trouble vasculaire peu commun qui commence généralement dans l’enfance, est plus fréquent chez les femmes et présente des signes de dépendance aux œstrogènes. L’angiome serpiginosum se caractérise par de multiples macules rouges-purpuriques asymptomatiques disposées en petits groupes suivant un motif serpiginosum le long des extrémités.
Le purpura prurigineux de Loewenthal,37 qui n’a été décrit que chez l’adulte, est considéré comme une variante plus symptomatique de la maladie de Schamberg.
Le PPD granulomateux, décrit par Saito,38 est une forme histopathologique plus fréquente chez les femmes et ne se distingue pas cliniquement des autres PPD.
Enfin, des formes familiales autosomiques dominantes de la maladie de Schamberg et du purpura annularis telangiectoides ont été rapportées39.
Histopathologie
Histopathologiquement, les PPD sont caractérisées par un infiltrat lymphocytaire périvasculaire centré sur les petits vaisseaux superficiels. Les autres observations typiques sont un gonflement endothélial, un rétrécissement de la lumière10, des GR extravasés et des macrophages chargés d’hémosidérine (Fig. 5 A et B). La coloration de Perls et Fontana-Masson montre des dépôts d’hémosidérine (fer) dans le derme superficiel, ce qui distingue la DPP de la dermatite de stase, qui présente des dépôts plus profonds.2
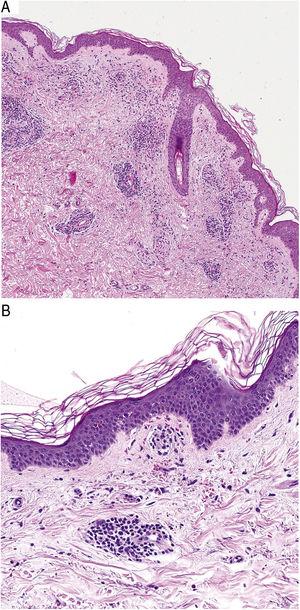
Caractéristiques histopathologiques de la maladie de Schamberg. A, Infiltrat impliquant des petits vaisseaux dans le derme superficiel. B, infiltrat lymphocytaire, avec rétrécissement luminal et globules rouges extravasés.
Un résultat caractéristique du lichen aureus est un épiderme intact séparé d’un infiltrat dermique en bande par une zone de tissu conjonctif épargné (zone de Grenz)27 (figure 6). Cet infiltrat est également typique de la dermatose lichénoïde purpurique pigmentée de Gougerot et Blum.26 Le purpura eczématoïde de Doucas et Kapetanakis, par contre, présente une spongiose épidermique et des neutrophiles dans l’infiltrat.25 La PPD granulomateuse se caractérise par un infiltrat granulomateux périvasculaire recouvrant des caractéristiques typiques.38 Une comparaison des caractéristiques cliniques, de la localisation et des résultats histopathologiques des variantes les plus courantes de la PPD est fournie dans le tableau 2.
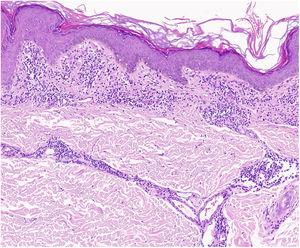
Caractéristiques histopathologiques du lichen aureus. Infiltrat en bande dans le derme papillaire et infiltrat périvasculaire superficiel.
Caractéristiques cliniques et histopathologiques des dermatoses purpuriques pigmentées.
| Variante | Présentation clinique | Localisation | Les résultats histopathologiques | |
|---|---|---|---|---|
| Maladie de Schamberg | Macules rouges-orange avec des taches périphériques ressemblant à des grains de poivre de Cayenne | Les membres inférieurs et parfois le tronc, bras, cuisses et fesses | Infiltrat lymphocytaire impliquant les petits vaisseaux superficiels, globules rouges extravasés, et des macrophages chargés d’hémosidérinemacrophages chargés d’hémosidérine | |
| Purpura eczématoïde de Doucas et Kapetanakis | Similaire aux manifestations de la maladie de Schamberg mais avec une desquamation et des démangeaisons intenses | Membres inférieurs | Infiltrat avec un plus grand nombre de neutrophiles et une spongiose épidermique | |
| Dermatose lichénoïde purpurique pigmentée de Gougerot et Blum | Papules lichénoïdes violacées qui fusionnent pour former des plaques | Membres inférieurs | Infiltrat dermique de type bandelette | .comme un infiltrat dermique |
| Lichen aureus | Isolé et persistant, rouge-orange et des lésions purpuriques | Membres inférieurs | Epiderme inchangé et infiltration dermique de type bande.comme un infiltrat dermique avec une zone de Grenz | |
| Plaques s’étendant de façon périphérique avec des pointes télangiectasiques sur les bords et s’estompant dans la zone centrale | Membres inférieurs et le tronc | Identique à la maladie de Schamberg |
Alors que certains auteurs1,13 considèrent que la capillarite est une caractéristique déterminante des DPP, Ackerman40 ne croit pas que ce soit le cas car il y a une absence de fibrine dans la paroi luminale et de thrombi dans les lumines.
Diagnostic
En plus de la biopsie cutanée, un test sanguin est recommandé pour exclure une thrombocytopénie, une coagulation ou des troubles auto-immuns (anticorps antinucléaires, facteur rhumatoïde) et des infections chroniques (anti-HCV et anti-HBsAg).2
Diagnostic différentiel
Le diagnostic différentiel doit inclure d’autres conditions présentant des manifestations purpuriques impliquant les extrémités inférieures. Ces entités et leurs principales caractéristiques sont résumées dans le tableau 3.
Diagnostic différentiel pour les dermatoses purpuriques pigmentées.
| Entités cliniques. | Main characteristics |
|---|---|
| Médicament-réactions d’hypersensibilité | Utilisation récente du médicament causal |
| Carbamazépine, méprobamate, chlordiazépoxide, furosémide, nitroglycérine, vitamine B1 et 5-fluorouracile topique | |
| Dermatite de contact purpurique aux vêtements | Les lésions sont confinées aux zones de la peau en contact avec les vêtements ; démangeaisons intenses |
| La laine, les colorants | |
| Purpura de stase veineuse | Signes d’une insuffisance veineuse chronique : gonflement, varices, sensation de lourdeur, ulcères veineux |
| Dépôt d’hémosidérine dans le derme profond | |
| Purpura dû à une thrombopénie | Associé à la numération plaquettaire |
| Purpura sénile | Chez les patients âgés, le purpura peut être associé à l’utilisation d’antiplaquettaires, d’anticoagulants, ou à la prise de corticostéroïdes |
| Exanthème purpurique dû à une infection virale | Autres signes d’infection |
| Vascularite leucocytoclastique | Lésions purpuriques palpables | A l’histologie : nécrose fibrinoïde, gonflement endothélial et leucocytoclasie |
| Purpura de Schönlein-Henoch | Age, 3-15 ans ; purpura symétrique affectant les jambes et les fesses ; douleurs articulaires et abdominales | Sarcome de Kaposi | Affecte les patients âgés ou immunodéprimés | A l’histologie : cellules fusiformes dans le derme formant des luminescences vasculaires irrégulières |
| Mycosis fongoïde purpurique | Début > 1 an ; disséminé, profil monoclonal dans l’infiltrat et perte de CD7 |
Source : Sardana et al.2, Kim et al,25 et Risikesan et al.26.
Dermoscopie
La constatation dermoscopique la plus courante est un fond rouge cuivré diffus qui correspond histopathologiquement à l’infiltrat dermique lymphocytaire, aux GR extravasés et aux macrophages chargés d’hémosidérine41. D’autres observations comprennent des globules et des points rouges, qui peuvent être expliqués par l’extravasation des GR, l’augmentation du nombre de vaisseaux sanguins et la dilatation de ces vaisseaux42 (Fig. 7). Des points bruns sont observés chez près de 50 % des patients et correspondent à la disposition sphérique ou elliptique des mélanocytes et des mélanophages à la jonction dermoépidermique. Dans un tiers des cas, la dermoscopie montre un pseudo-réseau pigmenté qui correspond à la couche basale hyperpigmentée et à l’incontinentia pigmenti dans le derme papillaire. Les résultats dermoscopiques spécifiques rapportés pour le lichen aureus comprennent un fond rouge cuivré avec des points et des globules bruns et rouges, des points gris et un pseudo-réseau comprenant des lignes pigmentées interconnectées43 (Fig. 8).
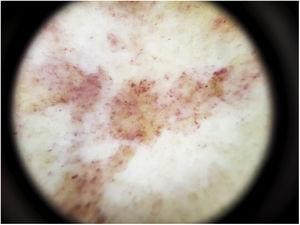
Caractéristiques dermoscopiques de la maladie de Schamberg. Fond rouge cuivré et globules rouges.
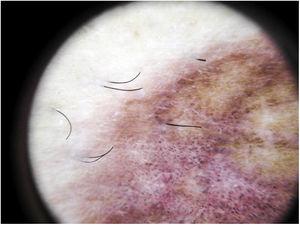
Manifestations dermoscopiques d’un lichen aureus. Fond brun-rouge avec des points gris et un pseudo-réseau pigmenté.
Traitement
Considérant que les DPP sont bénins et qu’il n’existe aucun traitement standardisé à l’efficacité prouvée, les risques et les avantages de tout traitement doivent être soigneusement pesés.
En raison de la nature bénigne et largement asymptomatique des DPP, aucun traitement n’est envisageable.4 Cependant, un traitement est souvent demandé en raison de la nature chronique de la maladie, de ses impacts physiques et psychologiques, et de la présence de lésions étendues ou de démangeaisons.
La plupart des recommandations de traitement sont basées sur de petites séries de cas, et aucune d’entre elles n’est étayée par des preuves suffisantes pour être considérée comme un traitement universel.
Une gamme de traitements topiques et systémiques, détaillés ci-dessous, ont été décrits dans de petites séries et des rapports de cas.
Traitements topiquesCorticostéroïdes topiques
Les corticostéroïdes topiques sont les traitements les plus couramment décrits et ont été observés pour réduire les démangeaisons et, dans certains cas, éclaircir les lésions2,4.
Les agents les plus utilisés sont les corticostéroïdes de moyenne et haute puissance (clobétasol et acéponate de méthylprednisolone)
Inhibiteurs de la calcineurine topiques
L’application topique de tacrolimus44 et de pimecrolimus45 pendant plusieurs mois s’est avérée résoudre le lichen aureus.
Vu la nature chronique des lésions de PPD et la nécessité d’un traitement à long terme, les inhibiteurs de la calcineurine peuvent être considérés comme une bonne alternative aux corticostéroïdes topiques pour éliminer ou résoudre les lésions.
La photothérapie
La photothérapie est une bonne option pour traiter une maladie étendue ou une PPD qui ne répond pas aux corticostéroïdes topiques ou aux inhibiteurs de la calcineurine.
Il a été postulé que la photothérapie peut être efficace car elle produit un effet immunomodulateur qui modifie l’activité des cellules T et réduit la production d’interleukine 2, ce qui entraîne une amélioration46.
Le traitement au psoralène et aux UV-A (PUVA) a été utilisé avec succès chez les patients atteints de la maladie de Schamberg, de dermatose lichénoïde purpurique et de lichen aureus. Dans les séries publiées à ce jour, entre 7 et 29 séances avec des doses cumulées allant de 16 à 49 J/cm2 ont été nécessaires pour obtenir une rémission. Un retraitement s’est également avéré efficace et, dans certains cas, un traitement d’entretien sur plusieurs mois a été nécessaire pour prolonger la réponse.2,47-49
La photothérapie UV-B à bande étroite avec des doses cumulatives comprises entre 11 et 49 J/cm2 administrées en 24 à 60 séances a produit des réponses favorables chez des patients présentant différentes variantes cliniques de la DPP. Comme pour le traitement par PUVA, des cas de récidive après l’arrêt du traitement ont été signalés, mais la réponse au retraitement était bonne.46,50,51
La photothérapie UV-B à bande étroite est considérée comme une bonne option en raison de son peu d’effets indésirables et de son bon profil de tolérance. Elle doit donc être considérée comme une option pour les patients pédiatriques, les patients présentant des lésions étendues et les patients résistants aux traitements topiques.5,52
Traitements systémiquesPentoxifylline
On a signalé des cas de DPP répondant à la pentoxifylline orale. Il a été suggéré que la pentoxifylline pourrait être efficace parce qu’elle inhibe l’adhésion des cellules T à l’endothélium vasculaire par interaction avec l’ICAM-1.53,54
La pentoxifylline a été utilisée seule, à une dose de 400 mg deux ou trois fois par jour pendant 2 à 3 mois,42,43 ou en association avec d’autres médicaments tels que les prostacyclines (prostaglandine I1)55 et les corticostéroïdes oraux56. La pentoxifylline s’est également avérée inefficace dans le traitement des PPD57.
Acide ascorbique et bioflavonoïdes (rutine/rutoside)Comme l’acide ascorbique et les bioflavonoïdes augmentent la production de collagène, réduisant ainsi la perméabilité vasculaire et améliorant la fonction de barrière endothéliale vasculaire, de fortes doses de vitamine C associées à un glycoside flavonoïde (comme le rutoside/rutine), présent dans les agrumes, administrées pendant plusieurs mois ont entraîné des améliorations cliniques et, dans certains cas, une résolution58.Autres traitements
Il existe des rapports isolés de réponse à divers traitements systémiques, tels que la griséofulvine,59 la colchicine,60 le méthotrexate,61 et la ciclosporine.62
Conclusions
Les DPP sont une affection dermatologique courante et ont un impact majeur sur la qualité de vie des patients en raison des symptômes et des préoccupations cosmétiques. Bien que les différentes variantes soient cliniquement très similaires, il existe un certain nombre de caractéristiques cliniques, histopathologiques et dermoscopiques qui aident à établir un diagnostic plus spécifique.
Enfin, bien qu’il n’y ait pas suffisamment de preuves dans la littérature pour recommander un traitement en première intention, il existe de nombreuses options qui permettent d’obtenir des améliorations considérables.
Conflits d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts.