Multifocal Motor Neuropathy (MMN) is a rare motor neuropathy with a reported prevalence range of 0.3-3 casos em 100.000,1 Afecta mais homens do que mulheres (2,7:1) e o seu início ocorre geralmente antes dos 50 anos de idade.1 A doença pode progredir para fraqueza e incapacidade permanentes, mas não é tão fatal ou incapacitante como a esclerose lateral amiotrófica (ALS, uma das doenças dos neurónios motores).2 Os sintomas MMN sobrepõem-se a outras doenças motor-predominantes, tais como a poliradiculoneuropatia desmielinizante crónica inflamatória (CIDP), variantes da ALS e da ALS (atrofia muscular progressiva, síndromes flail braço/perna flácida); esta sobreposição pode introduzir incerteza diagnóstica.
Reconhecer a sobreposição significativa na apresentação de MMN e ALS é particularmente importante dado que a MMN é tratável, enquanto que a ALS é rapidamente fatal e não tratável. O profundo impacto psicossocial de um diagnóstico de ELA está bem estabelecido e inclui luto, depressão, ansiedade, sentimentos de desespero e outros efeitos psicológicos negativos e alteradores da vida.3,4 Embora a ELA possa ser debilitante, não tem o mesmo prognóstico grave que a ELA e oferece a esperança de opções de tratamento.
ELA progride inexoravelmente, espalhando-se para envolver múltiplas funções motoras diferentes antes de eventualmente resultar em morte. O tempo médio de sobrevivência é de 3-5 anos e apenas cerca de 10% sobrevivem até 10 anos.5,6 As únicas opções de tratamento modificador da ALS são o riluzol e o edaravone.7-12 Em contraste, a MMN está associada a uma esperança de vida normal e tem várias opções de tratamento disponíveis,13-15 embora a MMN possa resultar em fraqueza muscular progressiva que pode levar a incapacidade grave se não for tratada.16 O tratamento precoce com imunoglobulina intravenosa (IVIg) é essencial para assegurar uma resposta óptima ao tratamento e evitar a progressão para a perda axonal.17 É fundamental, portanto, que a MMN seja correctamente diagnosticada o mais cedo possível, permitindo o início de uma terapia apropriada para prevenir efeitos permanentes, reduzir a incapacidade, e evitar o sofrimento psicológico de ser mal diagnosticada com ALS – uma doença uniformemente fatal. Infelizmente, o MMN pode ser muito difícil de diagnosticar em certos casos, particularmente cedo no curso da doença, e na ausência de bloqueio de condução óbvio (CB) e anticorpos GM1.
Este artigo visa discutir o diagnóstico diferencial de MMN e ALS, através de uma série de estudos de casos ilustrativos.
p>Características clínicas da neuropatia motora multifocal e da esclerose lateral amiotrófica
ALS é insidioso no início e afecta mais homens do que mulheres.1 A maioria das pessoas que desenvolvem a doença tem entre 40 e 70 anos, com uma idade média de 55 anos na altura do diagnóstico.18 No entanto, a ELA também ocorre em pessoas na casa dos vinte e poucos anos.1 Onset é assimétrico, com fraqueza a desenvolver-se numa região focal do rosto, braço ou perna. Os pacientes apresentam sinais de espasticidade, atrofia muscular rápida, enfraquecimento e desperdício. Isto leva a uma incapacidade de andar ou de mover os braços. O enfraquecimento muscular progride para os músculos do peito, levando, em última análise, a uma insuficiência respiratória. Tal como o MMN, a ALS pode apresentar uma queda do pé ou fraqueza da extremidade superior distal e atrofia, embora seja mais provável que envolva uma porção de um membro em vez de uma distribuição de um único nervo. Tal como na ALS, os pacientes com MMN podem manifestar atrofia muscular e fasciculações, embora as fasciculações sejam mais proeminentes na ALS (Tabela 1).1,19
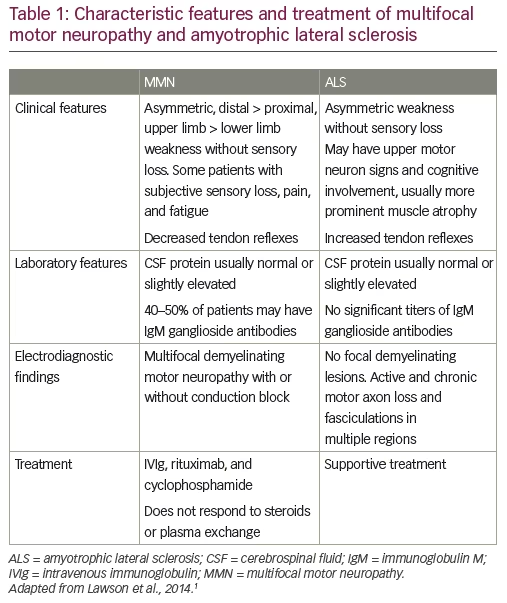
p>Embora o diagnóstico precoce de MMN possa ser difícil em casos atípicos, existem características clínicas cardinais que podem ajudar a estabelecer o diagnóstico. O MMN é caracterizado por fraqueza assimétrica dos membros sem perda sensorial, afectando mais frequentemente as extremidades superiores. A fraqueza é desigual e multifocal, correspondendo à distribuição de nervos únicos em vez de segmentar ou radicular (Tabela 1).1,20,21 Tal como a ALS, a doença tem um curso progressivo, mas a progressão da fraqueza tende a ser gradual, em vez de insidiosa. Os critérios clínicos fundamentais exigem o envolvimento de pelo menos dois nervos motores separados sem anomalias sensoriais objectivas, excepto no caso de uma ligeira deficiência do sentido vibratório. Um padrão electrofisiológico característico é a desaceleração focal e a CB das fibras nervosas motoras dentro dos segmentos nervosos. A estimulação da raiz nervosa cervical é também uma técnica importante para avaliar a CB, uma vez que aproximadamente 13% de todas as CB em MMN são proximais e podem falhar com estudos de rotina de condução nervosa.22,23
A ALS não comparável, que se acredita ser causada por uma combinação de factores genéticos e ambientais, o MMN tem claramente uma etiologia auto-imune; está associado a níveis elevados de anti-GM1 IgM em ~50% dos doentes e responde ao tratamento imunomodulador.21,24-26 A patofisiologia do MMN e do CB foi abordada noutros locais e não será revista aqui.24
Diagnóstico de neuropatia motora multifocal e esclerose lateral amiotrófica
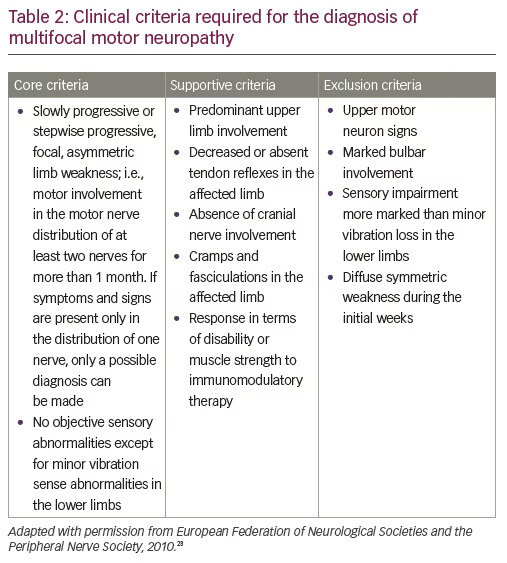
Um diagnóstico definitivo de MMN é baseado em critérios clínicos centrais estabelecidos pela Federação Europeia de Sociedades Neurológicas (EFNS)/Sociedade Nervosa Periférica (SNP) (critérios centrais, de apoio e de exclusão no diagnóstico de MMN são definidos pela EFNS; Quadro 2).28 Os critérios nucleares incluem fraqueza de membros lentamente progressiva ou gradual, focal, assimétrica, na distribuição de pelo menos dois nervos, durante mais de um mês; e nenhuma anomalia sensorial objectiva, excepto no caso de anomalias vibratórias menores nos membros inferiores.
As características importantes que excluiriam um diagnóstico de MMN incluem perda sensorial ou sintomas sensoriais que não sejam perda vibratória ligeira nos dedos dos pés, ou ligeira parestesia; bem como fraqueza simétrica no início.29 No entanto, estes seriam também atípicos para a ELA. Em termos de diferenciação da ELA e de outras perturbações neuropáticas motor-predominantes, outros critérios de exclusão para a ELA que ajudariam a distingui-la da ELA incluem os sinais de neurónios motores superiores e a fraqueza das barras. Os anticorpos IgM anti-ganglioside GM1 estão presentes em aproximadamente metade dos doentes com MMN (30-80% dependendo das séries).30,31 Contudo, também têm sido associados a outras neuropatias imuno-mediadas, neuropatias não relacionadas com a imunidade e mesmo doentes com ALS.32-34 Por conseguinte, os anticorpos anti-GM1 são diagnosticados mas não se pode confiar absolutamente.
A descoberta de CB motor também foi deixada fora dos critérios de diagnóstico do EFNS, uma vez que a CB em doentes com MMN típico pode não ser detectável utilizando testes electrofisiológicos clínicos padrão, um teste que requer conhecimentos electrofisiológicos consideráveis. A CB é definida como a falha de propagação potencial de acção num determinado local ao longo de um único axónio e a sua presença fora dos locais habituais de compressão nervosa nos testes de condução nervosa constitui a marca distintiva do MMN. Os critérios EFNS permitem um CB definitivo e provável.28 O CB definitivo é definido como se segue: “Pico negativo de potencial de acção muscular composto (CMAP) redução da área de estimulação proximal versus distal de pelo menos 50% independentemente do comprimento do segmento nervoso (mediana, ulnar, e peroneal). A amplitude do pico CMAP negativo na estimulação da parte distal do segmento com CB motor deve ser >20% do limite inferior do normal e >1 mV e o aumento da duração do pico CMAP negativo proximal a distal deve ser ≤30%”. O provável CB é definido por: “redução da área do pico CMAP negativo de pelo menos 30% num segmento longo (por exemplo pulso ao cotovelo ou cotovelo à axila) de um nervo do membro superior com aumento da duração do pico CMAP negativo proximal a distal ≤30%” ou “Redução da área do pico CMAP negativo de pelo menos 50% (o mesmo que definitivo) com um aumento da duração do pico CMAP negativo proximal a distal >30%”.28 Ao contrário da perda axonal, o CB será indetectável se localizado num local que não seja acessível através de testes convencionais de condução nervosa. A estimulação radicular proximal deve ser considerada nestes casos.22,23 Num estudo de doentes com doença pura dos neurónios motores inferiores sem CB motor, 10% responderam à terapia IVIg.35 Portanto, a ausência de anticorpos CB ou GM1 argumenta contra a reacção IVIg, mas não a exclui.
Além de CB e outras anomalias desmielinizantes, também pode haver evidência de perda de axónios motores, tais como diminuição do CMAP evocado distalmente e sinais de denervação e re-inervação na electromiografia com agulha.36 A neurografia por ressonância magnética é outra técnica de diagnóstico útil; a ampliação focal e o aumento da intensidade do sinal do plexo braquial é vista em imagens ponderadas em T2 no MMN.37 Além disso, muitos centros estão a utilizar ultra-sons para detectar a ampliação dos nervos; num estudo recente, a sonografia de alta resolução dos nervos periféricos revelou padrões distintos de ampliação multifocal do nervo, que podem apoiar um diagnóstico de MMN. Os resultados do ultra-som não se correlacionaram bem com a gravidade clínica ou os resultados electrofisiológicos na apresentação inicial, mas as alterações no Ultrasound Pattern Sum Score (UPSS) correlacionaram-se bem com o curso clínico em termos de força muscular, tal como medido pelo Medical Research Council sum score.38 O ultra-som tem sido utilizado para diferenciar o MMN de outras neuropatias39 e pode também ser uma ferramenta útil para a monitorização terapêutica.
Diagnóstico da ALS emprega os critérios de El Escorial e o algoritmo Awaji.40 De acordo com estes critérios, o diagnóstico da ALS requer sinais de degeneração de neurónios motores inferiores por exame clínico, electrofisiológico ou neuropatológico, e sinais de degeneração de neurónios motores superiores por exame clínico. Além disso, deve haver evidência de propagação progressiva de sinais dentro de uma região ou para outras regiões, ausência de evidência electrofisiológica de outros processos da doença, e ausência de evidência de neuroimagem de outros processos da doença que possam explicar os sinais clínicos e electrofisiológicos observados.
A sobreposição clínica significativa entre estes dois processos motores significa que o diagnóstico errado é comum e pode ser difícil de evitar. É importante identificar as características que ajudam a distinguir a MMN da ALS. Algumas destas diferenças já foram mencionadas anteriormente, mas merecem mais ênfase. Enquanto qualquer parte do corpo pode ser afectada na ALS, a MMN apresenta-se quase sempre com queda do pulso, ou menos frequentemente, queda do pé.17 Como tal, a MMN apresenta-se numa distribuição “desigual”, enquanto que a ALS envolve um segmento como um membro e espalha-se de forma insidiosa e não por etapas. O envolvimento de Bulbar e respiratório raramente é visto nas MMN, mas estão frequentemente presentes na ALS. A fraqueza muscular associada à MMN envolve menos atrofia, excepto em casos graves ou em indivíduos que têm a doença há muitos anos.41 Fasciculações ocorrem tanto na MMN como na ELA, mas são mais proeminentes e generalizadas na ELA. Além disso, na ALS, as fasciculações não se restringem necessariamente aos músculos fracos.1,19 A ausência de sinais de neurónios motores superiores é provavelmente um dos indicadores clínicos mais importantes da MMN sobre a ALS. No entanto, esta descoberta também deve ser interpretada com cautela, uma vez que existem variantes de neurónios motores inferiores da ALS que não são incomuns e também carecem de sinais de neurónios motores superiores (por exemplo, atrofia muscular progressiva). Electrofisiologicamente, a MMN distingue-se da ALS por CB, e por extensão, o recrutamento reduzido de unidades motoras na ausência de lesão axonal. O recrutamento reduzido é também observado na ALS mas é geralmente acompanhado por uma denervação mais proeminente na electromiografia com agulha.1,42
Diagnóstico de MMN ou ALS pode demorar mais de um ano, e um atraso no diagnóstico está associado a um pior prognóstico. Num estudo americano de 46 pacientes com MMN referidos a um centro neuromuscular terciário, apenas 6 receberam previamente o diagnóstico correcto.2 A proporção de MMN para ALS é de aproximadamente 1 a 20, e os pacientes com MMN são frequentemente diagnosticados como tendo ALS.41,43 O diagnóstico correcto do MMN requer frequentemente o envolvimento de um especialista neuromuscular com conhecimentos suficientes.
Tratamentos para neuropatia motora multifocal e evidências para o seu uso
As actuais directrizes EFNS/PNS recomendam o IVIg como a terapia padrão, baseada em evidências para MMN.44 Uma boa resposta ao IVIg é observada em até 80% dos doentes com MMN.21,45 A administração subcutânea de imunoglobulina também demonstrou eficácia no MMN e é mais conveniente do que a administração intravenosa.46-48 Outros desenvolvimentos como a administração facilitada por hialuronidase e formulações concentradas podem facilitar a administração subcutânea.49
Vários outros tratamentos incluindo ciclofosfamida, rituximab, micofenolato mofetil, beta-interferão, ciclosporina, azatioprina e infliximab foram todos utilizados para tratar MMN, mas dados insuficientes de ensaios clínicos apoiam a sua utilização para esta indicação.50 A troca de plasma ou corticosteróides são ineficazes ou prejudiciais no MMN, e a sua utilização deve ser evitada.51,52 As ciclosporinas de alta dose também mostraram alguma eficácia mas têm problemas de toxicidade, e os dados que apoiam a sua utilização são limitados.53 O Rituximab mostrou alguma eficácia em doentes com MMN, mas os dados são mistos e precisam de confirmação num grande ensaio clínico.54
As opções de tratamento para a ALS são mais limitadas. Actualmente são aprovados dois medicamentos que atrasam a progressão da ELA: o medicamento antiexcito tóxico riluzol, que está disponível há mais de 20 anos,8,9 e o recentemente aprovado edaravone, um antioxidante.55 Contudo, o edaravone só demonstrou eficácia num subconjunto de pacientes com ALS em fase inicial que satisfazem critérios específicos (ALS de grau 1 ou 2 na Classificação de Gravidade ALS do Japão, pontuação de pelo menos 2 pontos em todos os 12 itens da Escala de Classificação Funcional ALS Revista, capacidade vital forçada de 80% ou mais, ALS definida ou provável de acordo com os critérios revistos de El Escorial, e duração da doença de 2 anos ou menos).56
Estudos de caso
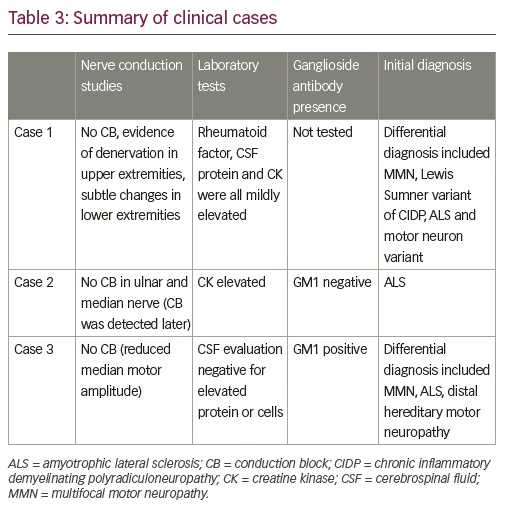
A série de casos que se segue são retirados da experiência dos autores e são típicos dos desafios em diferenciar a MMN da ALS (Tabela 3).
Caso 1
Um homem de 61 anos de idade apresentado 7 anos antes com uma fraqueza indolor da mão direita após uma cirurgia ao cotovelo. Com base na presença de fraqueza e atrofia, foi feito um diagnóstico clínico de neuropatia ulnar. Na reavaliação 2 anos mais tarde, tinha desenvolvido recorrência da fraqueza da mão direita sem perturbação sensorial. Embora sem dormência ou parestesia, relatou um ligeiro desconforto localizado à cintura do ombro na região subescapular que se estende aos rombóides e envolve o braço proximal. O desconforto diminuiu rapidamente, dando lugar a uma atrofia mais profunda da mão e do antebraço. A lesão do nervo interósseo posterior foi suspeita com base na fraqueza da extensão do dedo e punho, sem fraqueza da extensão do cotovelo associada ou alteração sensorial ao exame.
Um estudo de electrodiagnóstico realizado num hospital externo sugeriu lesão do nervo radial e neuropatia ulnar no cotovelo; o CB não foi identificado. Os outros membros não foram estudados. A impressão inicial ortopédica foi de uma neuropatia radial pós-traumática relacionada com uma lesão no cotovelo durante o treino de artes marciais. O paciente foi encaminhado para estudos neurológicos e de electrodiagnóstico repetido. Os estudos de condução nervosa revelaram respostas radiais, ulnares e motoras medianas de baixa amplitude à direita, com condução sensorial preservada, mas não preenchiam os critérios de CB. A electromiografia com agulha revelou potenciais de fibrilação proeminentes e ondas agudas positivas nos músculos distais da mão direita; denervação menos proeminente (tr-1+) nos músculos bíceps e tríceps com anomalias de recrutamento neurogénico associadas; potenciais de fibrilação esparsos e ondas agudas positivas no deltóide e no extensor digitorum communis da extremidade superior esquerda; e descargas repetitivas complexas nos músculos gastrocnémicos mediais sem evidência de denervação activa ou alterações crónicas da unidade motora. Suspeitou-se de uma desordem dos neurónios motores ou dos seus axónios com base nestes achados.
ALS foi o principal diagnóstico devido à ausência de CB definitivo; presença de denervação activa em múltiplos miotomos da extremidade superior direita e menos proeminente da extremidade superior esquerda; e alterações neurogénicas mais subtis nos músculos gastrocnémicos mediais das extremidades inferiores. As provas contra a ELA incluíram a ausência de sinais de neurónios motores superiores, a ausência de provas de denervação e re-inervação crónica, e a falta de progressão significativa ao longo de 2-3 anos.
Testes serológicos auxiliares revelaram um factor reumatóide ligeiramente elevado e creatina cinase sérica. A proteína do líquido cerebrospinal (LCR) também se encontrava ligeiramente elevada. O diagnóstico diferencial incluiu MMN, Lewis Sumner variante de CIDP, e ALS ou outra variante de neurónio motor.
A evolução da imunoglobulina foi iniciada a intervalos de 3 semanas após uma dose de carga de 2 g/kg durante 5 dias. Isto resultou numa melhoria clínica com aumento da força da extremidade superior direita e restauração gradual da massa muscular na mão direita.
Este caso realça algumas dificuldades importantes em distinguir a MMN da doença dos neurónios motores. Em particular, a ausência de CB definida pode tornar o diagnóstico essencialmente indistinguível de uma variante inferior do neurónio motor da ALS. A detecção de CB requer proficiência técnica, incluindo atenção às medições de distância, estimulação supramaximal, e colocação óptima dos eléctrodos. Mesmo com proficiência técnica, o CB pode não preencher critérios considerados “definitivos”, ou pode ser próximo de locais de registo. Isto sublinha a importância da electromiografia com agulha como parte do diagnóstico electrofisiológico. Em particular, as anomalias de recrutamento sem activos (potenciais de fibrilação e ondas agudas positivas) ou de denervação crónica podem ser uma pista importante.
A ausência de progressão significativa é um aspecto adicional importante do diagnóstico; a ELA progride inevitavelmente, e o diagnóstico é confirmado se um paciente desenvolver características definidoras, tais como disfunção bulbar ou sinais de neurónios motores superiores.
Caso 2
O paciente é um homem de 70 anos de idade diagnosticado com ELA após desenvolver fraqueza bilateral da mão que o deixou incapaz de trabalhar como escultor. As suas queixas iniciais eram de fraqueza da mão direita, como se manifestava pela perda de destreza e depois rápida perda de massa muscular na mão direita e no antebraço. A isto seguiu-se a fraqueza da extremidade inferior direita com queda do pé. Seguiu-se a fraqueza da extremidade distal inferior esquerda, mas manteve-se mais suave do que à direita. O CB não foi detectado no estudo inicial de electrodiagnóstico; apenas dois nervos motores foram estudados (nervo ulnar e nervo mediano à direita). Devido a estes sintomas e à ausência de CB no estudo electrodiagnóstico, foi-lhe dado um diagnóstico de ALS.
O paciente foi encaminhado para outra opinião quando não conseguiu progredir como era esperado para o seu diagnóstico de ALS. No momento da sua reavaliação, três dos seus quatro membros foram afectados. A fraqueza afectou mais proeminentemente os seus extensores de dedos, os intrínsecos das mãos, e o rapto do polegar do lado direito. Os grupos musculares afectados foram desperdiçados, mas não houve perda sensorial. A sua fala era clara, sem disartria ou disfonia. A língua era normal sem fasciculações. Os reflexos de estiramento muscular foram deprimidos por todo o lado. A presença de cãibras musculares, mais proeminentemente nas pernas e no tronco, provocou a ressonância magnética da medula torácica, que não era reveladora para o aumento da medula espinal ou da raiz. Estudos serológicos revelaram a elevação da creatina cinase em duas ocasiões distintas. Os títulos de ganglioside foram negativos para o anticorpo GM1. O diagnóstico de MMN foi confirmado pela descoberta de CB em locais nãopressivos no nervo ulnar direito, radial, e peroneal. Houve alguma denervação activa na tibialis anterior e no gastrocnémio medial, mas esta foi modesta e não foi acompanhada por provas de alterações significativas da unidade motora. Outros músculos estudados eram normais, à excepção de pronunciadas anomalias de recrutamento nos músculos radiais e ulnar-innerados sem evidência de denervação activa.
IVIg foi iniciado e resultou na melhoria da fraqueza em todas as extremidades. A força da extremidade superior direita melhorou mas não regressou à linha de base. Foi realizada uma avaliação da dependência de imunoglobulina (Ig) ~8 meses após o início da IVIg regular e tinha-se deteriorado em relação à força tanto nas extremidades superiores como inferiores com recidiva da queda do pé da extremidade inferior esquerda. A IVIg regular foi retomada e resultou num retorno à força observada antes do teste de dependência de Ig.
Este caso destaca o papel da reavaliação na avaliação de pacientes com ALS atípica. A reavaliação foi desencadeada quando o paciente não progrediu como esperado para a sua doença. As características atípicas incluíram a ausência de sintomas de bulbar e a ausência de sinais de neurónios motores superiores com reflexos difusamente deprimidos.
Na reavaliação, o paciente tinha progredido para envolver múltiplos nervos motores, e a evidência de CB nos nervos motores ulnar e radial cingiu o diagnóstico. A presença de mononeuropatias múltiplas pode imitar a distribuição miotomal da ELA na apresentação. Isto também realça a importância de manter um índice de suspeita no início da doença para detectar um padrão de fraqueza gradual sugestivo de uma mononeuropatia multifocal em vez de uma distribuição miotomal de fraqueza. A ausência de denervação na electromiografia com agulha deve ser uma importante pista de diagnóstico para um diagnóstico alternativo, e as anomalias desproporcionadas de recrutamento encaixam no MMN. Deve notar-se que no MMN, a CB proximal pode não ser detectável por estudos de condução nervosa, mas pode ser apreciada por anomalias de recrutamento. Além disso, este caso realça a importância de uma amostragem adequada dos nervos motores precoce com atenção à realização de estímulos proximais.
Case 3
Um homem de 52 anos de idade apresentado à clínica ortopédica com queixas de “o seu dedo anelar direito ficar preso” após a utilização de tesouras por um período prolongado. A sua fraqueza tinha progredido ao longo dos 12-18 meses anteriores. Ao rever a sua história, admitiu que a progressão foi “intermitente”, relatando um agravamento abrupto da força das mãos nos 4-6 meses que antecederam a apresentação. Teve dificuldades com a flexão dos 4º e 5º dígitos da mão direita e manteve o seu 5º dígito numa posição raptada. Não conseguiu segurar o seu 5º dígito em toda a extensão e teve fraqueza de raptos dos dedos. Não houve dor associada, dormência ou formigueiro, mas queixou-se de cólicas localizadas nos braços, costas e ombros.
Foi encaminhado para consulta neurológica e avaliação electrodiagnóstica devido à atrofia hipotenar; foi sugerido um diagnóstico putativo de neuropatia ulnar. O exame neurológico revelou nervos cranianos normais sem evidência de disfunção de bulbar, atrofia da língua ou fasciculações. Ao exame motor, apresentava uma ligeira atrofia dos músculos do entãoar direito, hipotenar e do antebraço. Tinha uma ligeira atrofia esquerda do entãoar. O teste de força revelou 4-/5 de rapto do dedo direito e 5-/5 na extensão do dedo; 4-/5 de rapto do polegar esquerdo; e 4/5 de dorsiflexão do tornozelo direito e do dedo do pé. Rapto do ombro, flexão do cotovelo e resistência da extensão do cotovelo foram normais bilateralmente, tal como a resistência da extremidade inferior proximal. Os reflexos eram 3+ na patela, ausentes no tornozelo direito, 2+ no tornozelo esquerdo, e diminuíram nas extremidades superiores bilaterais. O estudo de electrodiagnóstico não conseguiu identificar o CB, mas a amplitude de resposta motora mediana direita foi reduzida. A denervação estava presente no primeiro interósseo dorsal, mas não era proeminente. Não houve alterações de unidades motoras tais como polifásia, grande amplitude ou unidades de longa duração.
Anticorpos gangliosídicos revelaram a positividade de GM1 a um nível elevado. A avaliação do LCR foi negativa para proteínas ou células elevadas. Com base na presença de anomalias de recrutamento proeminentes, positividade GM1 e um historial de mudança gradual na função da mão, foi concluído um ensaio de IVIg. Uma avaliação neurológica repetida ~6 meses após a terapia com Ig revelou uma melhoria na força de abdução do polegar à esquerda e dorsiflexão do tornozelo à direita. O rapto do dedo direito também foi melhorado, mas em menor grau. Suspeitou-se de um diagnóstico de MMN com base nesta resposta e ele permaneceu em IVIg.
Perspectivas futuras
Como ilustrado por estes exemplos de casos, há necessidade de mais técnicas para distinguir ALS de MMN. Verificou-se que as duas condições exibem perfis distintos de citocinas e quimiocinas em doentes. Um estudo de 2015 (n = 56) encontrou diferenças nas características inflamatórias do LCR entre pacientes com MMN e aqueles com ELA; em particular, os níveis do factor de crescimento do fibroblasto-2 e do factor de estimulação da colónia de granulócitos foram elevados em pacientes com ELA em comparação com aqueles com MMN.57
Além disso, como discutido anteriormente, dados recentes mostraram que o ultra-som de nervos tem uma elevada precisão diagnóstica no diagnóstico diferencial de ELA e MMN, e pode ser superior aos estudos de condução nervosa no diagnóstico de MMN em pacientes hospitalizados com este diagnóstico diferencial.38,58 Outro estudo recente sugeriu que a ecografia cervical de raiz pode ser uma técnica útil para apoiar o diagnóstico de MMN em vez de ALS, mesmo na ausência de CB.59 Um diagnóstico mais rápido e preciso pode levar a um maior número de pacientes a receberem IVIg precoce ou outros tratamentos de MMN, embora o resultado a longo prazo de um diagnóstico precoce mais generalizado permaneça desconhecido.
Sumário e observações finais
p> O diagnóstico errado de MMN como ALS é uma questão importante com sérias implicações clínicas para o paciente devido a diferenças no prognóstico e tratamento. Nesta revisão, foram discutidas algumas características clínicas importantes que podem ajudar a distinguir as duas doenças. Em resumo, as seguintes características devem alertar o médico para um possível diagnóstico de MMN: (i) envolvimento distal do membro superior (embora o médico também deva estar ciente de que se trata de um sintoma comum da ELA); (ii) progressão multifocal e gradual na distribuição de nervos únicos; (iii) ausência de envolvimento bulbar/respiratório; (iv) anormalidades CB/demyelinizantes no estudo electrofisiológico; (v) ausência de sinais de neurónios motores superiores; (vi) fasciculações esparsas e (vii) anticorpos GM1.1,20,21,29
No entanto, é preciso repetir que as variantes de ALS sem sinais de neurónios motores superiores podem ser extremamente difíceis de distinguir de síndromes de neurónios motores inferiores imunes, tais como MMN. A presença de anticorpos GM1 ou CB deve sempre levar à consideração de um ensaio IVIg; dado que a maioria dos doentes responderá em 8-12 semanas de doses de reforço, isto é apropriado dada a gravidade de um diagnóstico errado. Do mesmo modo, em doentes com síndromes de neurónios motores inferiores puros sem anticorpos CB ou GM1, deve ser considerado um ensaio de IVIg.
É necessário divulgar informação sobre o diagnóstico clínico de MMN. As apresentações acima referidas de casos do “mundo real” demonstram como o diagnóstico incorrecto pode ser evitado. Considerações importantes são a necessidade de conhecimentos técnicos especializados em testes de electrodiagnóstico, uma vez que o CB definitivo nem sempre está presente, e a necessidade de avaliações neurológicas repetidas. Quando correctamente diagnosticada, a MMN responde frequentemente bem à IVIg. Contudo, o diagnóstico atrasado prejudicará a eficácia do tratamento e pode resultar numa diminuição da força muscular, incapacidade e pior prognóstico.