R. Kishore Kumar e Surinder Paul Negi*
Do Departamento de Pediatria e *Medicina de Emergência, North West Regional Hospitaland University of Tasmania, Austrália.
Reimpressão de pedidos: Dr. R. Kishore Kumar, Consultor Pediatra e Lecutador em Pediatria e Saúde Infantil, North West Regional Hospital and University ofTasmania, P O Box 258, Burnie TAS 7320, Austrália. E-mail: [email protected]
ManuscritoRecebido: 5 de Abril de 1999;
Revisão inicial concluída: 26 de Maio de 1999;
RevisãoAceitada: 18 de Agosto de 1999
Síndrome da triploidia é uma síndrome rara e estima-se que ocorra em cerca de 2 por cento dos conceitos(1). A identificação da síndrome é da maior importância, uma vez que a maioria morre nos primeiros 2 meses, e também para a família, para o aconselhamento genético posterior, planeamento para a família futura e para o seu luto. Relatamos um caso para destacar estas questões.
Case Report
Um recém-nascido foi transferido para o nosso hospital a partir de um centro periférico para uma avaliação mais aprofundada devido ao “aparecimento invulgar”. O bebé foi o segundo filho do casal caucasiano não linguístico. A mãe tinha 30 anos de idade. Eles têm um menino de 3 anos de idade, que é saudável e está bem. A segunda gravidez tinha sido abortada espontaneamente às 14 semanas de gestação. Não havia historial familiar de quaisquer doenças significativas ou quaisquer outras perturbações. O bebé era pequeno para a idade gestacional com grave deficiência de crescimento intra-uterino com um peso ao nascer de 1600 g às 41 semanas de gestação. Tinha uma grande fontanela anterior e uma fontanela posterior, orelhas baixas, hipertelorismo, nariz pontiagudo e um palato arqueado alto. Havia sindactilia de terceiro e quarto dígitos, tanto nos membros superiores como nos inferiores. O vinco simiano foi notado em ambas as mãos. Os testes não eram descendentes em ambos os lados e o comprimento do pénis era inferior a 0,5 cm. Apenas 2 vasos umbilicais foram anotados. Tinha também pés de balancim em ambos os lados. Também teve um pequeno defeito do septo ventricular confirmado no ecocardiograma bidimensional. Tendo em conta o acima exposto, suspeitou-se que ele tivesse trissomia 18. A sua ecografia renal foi relatada como sendo normal. O seu estudo cromossómico revelou uma deleção de 18p, mas, de resto, nenhuma evi-dência de qualquer trissomia. Em todas as células analisadas, um homólogo do cromossoma 18 pareceu atípico dentro da região centrrométrica do braço curto. Em virtude disto, foi realizado um estudo cromossómico parental revelando cromossomas maternos com deleção de 18p, exactamente o mesmo que o bebé. Mas a mãe é fenotípicamente normal, sem evidência de dismorfismo e sem deficiência mental. Por conseguinte, isto foi atribuído pelos geneticistas como sendo um achado incidental. O bebé foi analisado pelo nosso geneticista clínico sénior, que diagnosticou a síndrome da triploidia e foi realizada uma biópsia da pele. A cultura do fibroblasto cutâneo revelou cariotipagem do mosaico 46XY/69XXY, consistente com o diagnóstico da síndrome da triploidia do mosaico.
Desde que o nosso caso é uma triploidia do mosaico, o bebé ainda está vivo aos 7 meses de idade, pesando apenas 3,17 kg, com um atraso de desenvolvimento acentuado. Os pais aceitaram a realidade e aceitaram a realidade. Desde o diagnóstico, o número de atendimentos a vítimas de “problemas de alimentação e de não ganhar peso” tem sido nulo, em comparação com 10 visitas nas primeiras 8 semanas ao médico de clínica geral. A família, consciente de que o risco de recorrência é insignificante, espera o seu terceiro filho com muito pouca apreensão, embora esteja a planear ter amiocentese às 16 semanas de gestação para confirmar isto. O diagnóstico deste bebé com síndrome da triploidia, fez com que a
família chegasse a um acordo e tivesse tempo suficiente para lamentar, planear o seu próximo filho de uma forma muito descontraída; ajudou o médico de clínica geral a aconselhar adequadamente a família e o pediatra a organizar um aconselhamento apropriado para o seu próximo filho.
Discussão
Síndrome da triploidia é caracterizada por dismaturidade geral, hipotonia muscular, grande fontanela posterior, aurículas dismórficas de baixo set, hipertelorismo, microftalmia e colobomata, sindactilia cutânea do terceiro e quarto dedos, sulco símio, hipospadias e/ou genitália externa mal desenvolvida(2). A maioria dos bebés nascem prematuros e normalmente vivem menos de um dia. Pode haver evidência de uma grande placenta com frequentes alterações hidatidiformes. Todos os casos de triploidia completa ou nasceram natimortos ou morreram no período neonatal precoce(3), sendo cinco meses a maior sobrevivência registada(4).
A importância de um diagnóstico confirmado correcto para qualquer criança, em particular para uma família que se depara com um bebé “dismórfico” e que está a planear ter mais filhos não pode ser sobrestimada. As doenças sindrómicas envolvendo trissomias de cromossomas são facilmente reconhecidas pelos pediatras. A questão surge quando os cromossomas são relatados como normais, quando as pessoas assumem que se trata de um problema “idiopático”. A síndrome da triploidia é uma dessas síndromes, em que as linhas celulares triplóides podem ter desaparecido do sangue periférico e a evidência da triploidia só pode ser encontrada nos fibroblastos cutâneos cultivados. A síndrome de Palliester Killian é uma dessas síndromes onde na tetrasomia 12p foi documentada em fibroblastos cutâneos de indivíduos afectados, mas não a partir do sangue periférico. Na maioria dos casos de síndrome da triploidia, o conjunto extra de cromossomas é paternalmente derivado, com 66% atribuídos à fertilização dupla, 24% devido à fertilização com um esperma diplóide e
10% como fertilização de um óvulo diplóide. A idade materna mais velha não tem sido um factor diferente da maioria das trissomias. Não há dados que indiquem um risco acrescido, como os observados com a não disjunção cromo-somal, o que significa que se pode ser mais optimista ao mesmo tempo que se oferece aconselhamento genético para futuras crianças. Mas em vários casos, uma gravidez triplóide é seguida ou precedida por uma gravidez molar.
Tem de se pensar nesta possibilidade mais cedo do que mais tarde, tendo em conta a reduzida esperança de vida destes bebés, para que possam fazer testes apropriados para oferecer aconselhamento genético à família envolvida, de modo a dar-lhes tempo suficiente para o luto e para se adaptarem à realidade e também para planearem novas gravidezes sem serem apreensivos. Uma pesquisa bibliográfica revelou apenas 17 citações a este tópico no mundo até agora, sendo que a sobrevivência mais longa foi de 5 meses. Relatamos este caso para destacar a sua existência e a sobrevivência prolongada do nosso caso provavelmente devido ao mosaicismo.
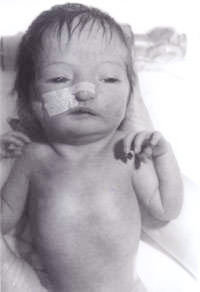
Fig. 1. Bebé com síndrome da triploidia.
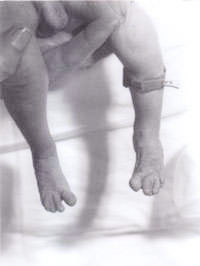
Fig. 2. Sindactalia do terceiro e quarto dedos do pé.
Agradecimentos
Os autores gostariam de agradecer à Dra. Agnes Bankier, Geneticista Clínica Sénior e ao Dr. Stephen Robertson, Fellow in Clinical Genetics at Royal Children’s Hospital, Melbourne, pela sua ajuda no diagnóstico e aconselhamento genético da família.
1. Jones KL. Síndrome da triploidia e síndrome da mixoploidia diplóide/triploidia. In: Smith’s Recognizable Patterns of Human Malformation, 4th edn. Philadelphia, 1988; pp 32-33.
2. Leisti JT, Raivio KO, Rapola MH, Saksela EJ, Aula PP. O fenótipo da triploidia humana. Os Defeitos de Nascimento Origem Artic Ser 1974; 10: 248-253.
3. Garzena E, Farinasso D, Prandi GM, Vardeu P, Bagna R, Cavo L, et al. Síndrome da triploidia: Um relato de caso. Minerva Pediatr 1995; 47: 307-311.
4. Arvidsson CG. Um rapaz com uma tripolidia completa e uma sobrevivência invulgarmente longa. Acta Paediatr Scand 1986; 75: 507-510.