Introduzione
Le dermatosi purpuriche pigmentate (PPD) sono un raro gruppo di malattie benigne croniche caratterizzate da petecchie multiple su macule iperpigmentate, giallo-marroni.1 Le diverse varianti sono forme cliniche distinte della stessa entità con caratteristiche istopatologiche simili.2,3
Ci sono 5 varianti classiche: Malattia di Schamberg (PPD progressiva), porpora eczematoide di Doucas e Kapetanakis (porpora pruritica), dermatosi lichenoide purpurica pigmentata di Gougerot e Blum, lichen aureo (lichen purpuricus) e malattia di Majocchi (porpora annularis telangiectodes).1 Varianti meno comuni sono la PPD granulomatosa, la porpora prurito di Loewenthal, la PPD lineare, la PPD transitoria e la PPD familiare.2
Epidemiologia
Le PPD sono rare e colpiscono prevalentemente gli adulti,1 anche se sono stati riportati casi nei bambini.4 La variante più comune sia negli adulti che nei bambini è la malattia di Schamberg. Le PPD, e le forme lineari in particolare,6 sono generalmente più comuni negli uomini.1 La malattia di Majocchi è più comune nelle donne.
Eziologia e patogenesi
Anche se le cause delle PPD sono sconosciute, è stata proposta una serie di fattori scatenanti, tra cui esercizio fisico, ipertensione venosa, diabete mellito, infezioni,2,7 e vari farmaci (Tabella 1).8 Un’associazione con dislipidemia e malattie autoimmuni è stata riportata per la PPD granulomatosa.9 Nella maggior parte dei casi, tuttavia, non viene identificata alcuna causa.10
Farmaci associati alle dermatosi purpuriche pigmentate.
| Sedativi | Fenobarbital, clordiazepossido, meprobamato | |
| Vitamine | Tiamina (B1) | |
| Diuretici | Furosemide | |
| Farmaci cardiovascolari | Nitroglicerina, bezafibrato, idralazina, dipiridamolo, sildenafil | |
| Antibiotici | Ampicillina | |
| Analgesici | Farmaci antinfiammatori non steroidei, aspirina, acetaminofenolo | |
| Stimolanti | Pseudoefedrina | |
| Ormoni | Medrossiprogesterone acetato | |
| Farmaci anti-diabetici | Glipizide | |
| Agenti chemioterapici | Topico 5-fluorouracile | |
| Antivirali | Interferone α | |
| Retinoidi | Isotretinoina |
Fonte: Kaplan et al.8
Alla dilatazione e fragilità capillare è stato attribuito un possibile ruolo patogeno nei PPD.10 È stato ipotizzato che le cellule responsabili di questi disturbi siano cellule coinvolte nella struttura dei vasi sanguigni, come i fibroblasti e le cellule endoteliali. Sia attraverso l’attivazione (ad esempio, alta pressione intravascolare) o spontaneamente, la funzione di queste cellule può essere alterata, causando la fuoriuscita di globuli rossi (RBC) attraverso le pareti dei vasi,11 innescando una reazione di ipersensibilità cellulo-mediata. La risposta immunitaria cellulo-mediata sembrerebbe quindi avere un ruolo fondamentale nella patogenesi delle PPD.12,13 L’infiltrato infiammatorio perivascolare è composto da cellule T CD4+14 (con ridotta espressione di CD715) e cellule dendritiche CD1a+.12
Il ruolo patogenetico delle molecole di adesione cellulare nelle PPD è stato anche analizzato in diversi studi. Le CAM sono proteine di membrana che interagiscono con ligandi specifici che forniscono e mantengono il contatto tra cellule diverse e tra cellule e proteine della matrice extracellulare. Sono stati osservati alti livelli di espressione per le molecole di adesione LFA-1 (antigene funzionale dei linfociti-1) e ICAM-1 (molecola di adesione intercellulare-1) nelle cellule infiammatorie e per ICAM-1 e ELAM-1 (molecola di adesione dei leucociti endoteliali – 1)12 nelle cellule endoteliali. Le cellule T attivate da uno stimolo antigenico aderirebbero così alle cellule endoteliali, ai fibroblasti e ai cheratinociti.16 Le citochine prodotte dai leucociti (per esempio, il fattore di necrosi tumorale α) possono innescare l’espressione di queste molecole di adesione (Fig. 1).
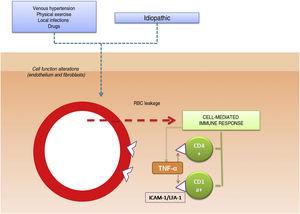 Meccanismo eziologico e patogenetico. Una delle ipotesi più ampiamente accettate è che le cellule T sono attivate da uno stimolo antigenico e si legano a cellule endoteliali, fibroblasti e cheratinociti attraverso l’espressione di molecole di adesione. TNF-α indica il fattore di necrosi tumorale α; ICAM-1, molecola di adesione intercellulare-1; LFA-1, antigene di funzione linfocitaria-1.
Meccanismo eziologico e patogenetico. Una delle ipotesi più ampiamente accettate è che le cellule T sono attivate da uno stimolo antigenico e si legano a cellule endoteliali, fibroblasti e cheratinociti attraverso l’espressione di molecole di adesione. TNF-α indica il fattore di necrosi tumorale α; ICAM-1, molecola di adesione intercellulare-1; LFA-1, antigene di funzione linfocitaria-1.Meccanismo eziologico e patogenetico. Una delle ipotesi più ampiamente accettate è che le cellule T sono attivate da uno stimolo antigenico e si legano a cellule endoteliali, fibroblasti e cheratinociti attraverso l’espressione di molecole di adesione. TNF-α indica il fattore di necrosi tumorale α; ICAM-1, molecola di adesione intercellulare-1; LFA-1, antigene di funzione linfocitaria-1.
Le suddette citochine possono anche provocare una diminuzione del rilascio dell’attivatore del plasminogeno endoteliale e/o un aumento eccessivo dell’inibitore dell’attivatore del plasminogeno,17 portando alla ridotta attività fibrinolitica e al deposito intraperivascolare di fibrina osservato nelle PPD.18
L’immunofluorescenza diretta può mostrare la deposizione di fibrinogeno, immunoglobulina M e/o C3 nei vasi dermici superficiali.10
Un’altra ipotesi emersa negli ultimi anni è che le PPD possano rappresentare un’insidiosa alterazione epiteliotropa delle cellule T. Questa teoria è supportata dall’osservazione dell’epidermotropismo o di un pattern monoclonale nell’infiltrato infiammatorio.15,19 Ci sono state anche alcune segnalazioni di progressione a micosi fungoide.20-22 Poiché è difficile distinguere tra micosi fungoide purpurea e PPD monoclonale, è essenziale integrare i risultati clinici, molecolari e istopatologici.15,20,23 Poikiloderma, prurito, placche coalescenti, una durata superiore a 1 anno, un pattern monoclonale e una ridotta espressione di CD7 e CD62 L nell’infiltrato dovrebbero far sospettare una progressione della malattia, anche in assenza di atipia linfocitaria evidente.15,24 Alcuni autori scelgono di trattare la DPP disseminata e monoclonale come micosi fungoide allo stadio iniziale.
Varianti clinicheLa PPD progressiva o malattia di Schamberg1,2
Nella PPD progressiva o malattia di Schamberg, le lesioni di solito appaiono su entrambe le estremità inferiori, ma possono anche interessare il tronco, le braccia, le cosce o le natiche. Si presentano come macule rosso-arancioni con macchie periferiche purpuriche simili a grani di pepe di Caienna (Fig. 2A e B); queste macchie acquisiscono un colore giallo-marrone man mano che progrediscono. Le lesioni sono generalmente asintomatiche, anche se alcuni pazienti descrivono un prurito. Seguono un decorso cronico con numerose ricadute e remissioni.
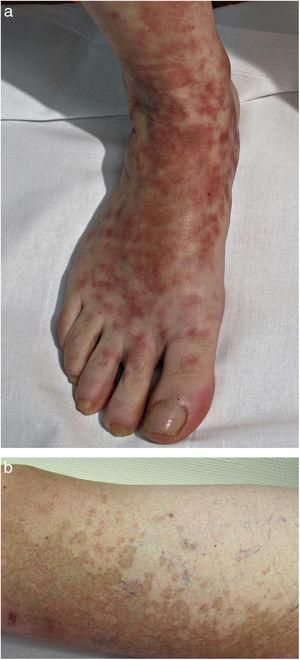
Malattia di Schamberg. A, macule rosso-arancio che tendono a coalizzarsi, formando grandi placche. B, macchie periferiche di pepe di cayenna.
La porpora purpurea o porpora eczematoide di Doucas e Kapetanakis1,25
La porpora purpurea o porpora eczematoide di Doucas e Kapetanakis è la variante più estesa e pruritica delle PPD. Colpisce soprattutto le estremità inferiori ed è clinicamente simile alla malattia di Schamberg, con macule purpuriche o petecchiali ma con una superficie desquamata. La porpora eczematoide di Doucas e Kapetanakis è stata associata a una dermatite allergica da contatto con gomma e indumenti. L’esordio è rapido (15-30 giorni) e le lesioni possono durare mesi o anni.
Dermatosi purpurica pigmentata lichenoide di Gougerot e Blum1,2,26
La dermatosi purpurica lichenoide di Gougerot e Blum è caratterizzata da papule lichenoidi violacee che tendono a fondersi, formando grandi placche che di solito sono localizzate sulle gambe ma possono interessare il tronco. La condizione segue un decorso cronico e di solito colpisce gli uomini anziani. Deve essere distinto dal sarcoma di Kaposi.
Lichen Aureus o Lichen Purpuricus1,2,27
Lichen aureus o lichen purpuricus è una variante più localizzata della PPD. Le lesioni sono persistenti e sono tipicamente solitarie o in numero ridotto. Il lichen aureo è caratterizzato dall’improvvisa comparsa di piccole papule giallo-arancioni con un aspetto lichenoide e una tendenza a coalizzarsi in placche che misurano tra 1 e 20 cm associate a lesioni purpuriche millimetriche (Fig. 3). La condizione colpisce soprattutto le estremità inferiori, ma le lesioni possono verificarsi su qualsiasi parte del corpo. Di solito sono asintomatiche. Nei bambini e negli adolescenti sono state descritte varianti zosteriformi28 e segmentali lungo le linee di Blaschko29 o che seguono il corso delle vene safene30 o cefaliche31.
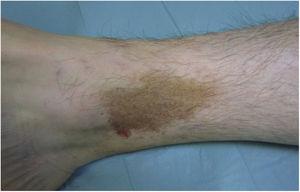 Lichen aureus. Placca solitaria composta da piccole macule giallo-arancio coalescenti sulla gamba.
Lichen aureus. Placca solitaria composta da piccole macule giallo-arancio coalescenti sulla gamba.Lichen aureus. Placca solitaria comprendente piccole macule giallo-arancio coalescenti sulla gamba.
Purpura Annularis Telangiectodes o malattia di Majocchi1,2,25
Purpura annularis telangiectodes o malattia di Majocchi si presenta con macule anulari rosso-violacee (Fig. 4), seguite da punti telangiectatici rosso più scuro. Le lesioni si estendono perifericamente e il loro centro svanisce gradualmente e può acquisire un aspetto atrofico. L’eruzione inizia nelle estremità inferiori e poi si diffonde al tronco e alle braccia; è caratterizzata da un gran numero di lesioni. È stata descritta una variante nota come porpora arciforme annularis telangiectodes, caratterizzata da un minor numero di lesioni, ma più grandi, con una caratteristica morfologia arcuata.32
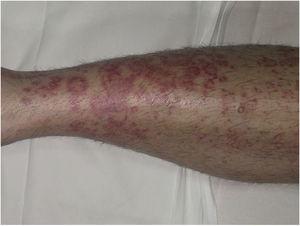 Malattia di Majocchi. Lesioni anulari rosso-violacee sulla gamba.
Malattia di Majocchi. Lesioni anulari rosso-violacee sulla gamba.Malattia di Majocchi. Lesioni anulari rosso-violacee sulla gamba.
Altre varianti
Hersch e Schwayder33 hanno descritto quella che è considerata una rara forma lineare e unilaterale che deve essere differenziata dalle forme lineari della malattia di Schamberg e dal lichen aureo. Higgins e Cox34 hanno descritto una forma quadrangolare che hanno attribuito a un’ostruzione vascolare nella pelvi.
Ci sono state anche segnalazioni di una variante transitoria35 che include entità come l’angioma serpiginosum,36 che è un disordine vascolare non comune che di solito inizia nell’infanzia, è più comune nelle femmine e mostra segni di dipendenza dagli estrogeni. L’angioma serpiginosum è caratterizzato da macule multiple asintomatiche rosso-purpuree disposte in piccoli gruppi secondo uno schema a serpiginosum lungo le estremità.
Purpura di Loewenthal,37 che è stata descritta solo negli adulti, è considerata una variante più sintomatica della malattia di Schamberg.
La PPD granulomatosa, descritta da Saito,38 è una forma istopatologica più comune nelle donne e clinicamente indistinguibile dalle altre DPP.
Infine, sono state segnalate forme familiari autosomiche dominanti della malattia di Schamberg e della porpora annularis telangiectoides.39
Istopatologia
Sotto il profilo istopatologico, le PPD sono caratterizzate da un infiltrato linfocitario perivascolare centrato sui piccoli vasi superficiali. Altri reperti tipici sono il rigonfiamento endoteliale, il restringimento del lume,10 i RBC extravasati e i macrofagi carichi di emosiderina (Fig. 5 A e B). La colorazione di Perls e Fontana-Masson mostra depositi di emosiderina (ferro) nel derma superficiale, distinguendo la DPP dalla dermatite da stasi, che ha depositi più profondi.2
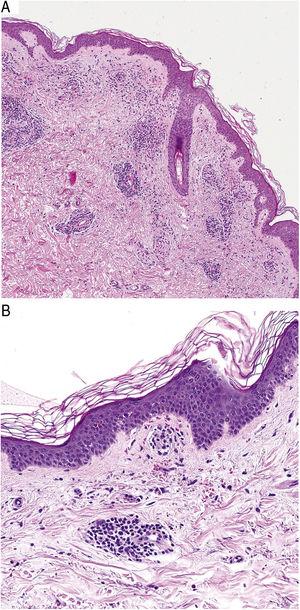 Caratteristiche istopatologiche della malattia di Schamberg. A, infiltrato che coinvolge piccoli vasi nel derma superficiale. B, infiltrato linfocitario, con restringimento luminale e globuli rossi extravasati.
Caratteristiche istopatologiche della malattia di Schamberg. A, infiltrato che coinvolge piccoli vasi nel derma superficiale. B, infiltrato linfocitario, con restringimento luminale e globuli rossi extravasati.Caratteristiche istopatologiche della malattia di Schamberg. A, infiltrato che coinvolge piccoli vasi nel derma superficiale. B, infiltrato linfocitario, con restringimento luminale e globuli rossi extravasati.
Un reperto caratteristico del lichen aureo è un’epidermide intatta separata da un infiltrato dermico a banda da un’area di tessuto connettivo risparmiato (area di Grenz)27 (Fig. 6). Questo infiltrato è tipico anche nella dermatosi lichenoide purpurica pigmentata di Gougerot e Blum.26 La porpora eczematoide di Doucas e Kapetanakis, invece, presenta spongiosi epidermica e neutrofili nell’infiltrato.25 La PPD granulomatosa è caratterizzata da un infiltrato granulomatoso perivascolare che si sovrappone alle caratteristiche tipiche.38 Un confronto tra le caratteristiche cliniche, la localizzazione e i risultati istopatologici per le varianti più comuni della PPD è fornito nella Tabella 2.
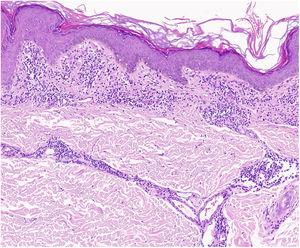 Caratteristiche istopatologiche del lichen aureus. Infiltrazione a banda nel derma papillare e infiltrato perivascolare superficiale.
Caratteristiche istopatologiche del lichen aureus. Infiltrazione a banda nel derma papillare e infiltrato perivascolare superficiale.Caratteristiche istopatologiche del lichen aureo. Infiltrazione a banda nel derma papillare e infiltrato perivascolare superficiale.
Caratteristiche cliniche e istopatologiche delle dermatosi purpuriche pigmentate.
| Variante | Presentazione clinica | Localizzazione | Riscontri istopatologici |
|---|---|---|---|
| Malattia di Schamberg | Macule rosso-macule arancioni con macchie periferiche simili a grani di pepe di cayenna | Arti inferiori e occasionalmente tronco, braccia, cosce e natiche | Infiltrato linfocitario che coinvolge i piccoli vasi superficiali, globuli rossi stravasi, ed emosiderinamacrofagi carichi di emosiderina |
| Purpura eczematoide di Doucas e Kapetanakis | Simile alle manifestazioni della malattia di Schamberg ma con desquamazione e prurito intenso | Arti inferiori | Infiltrato con più numero di neutrofili e spongiosi epidermica |
| Dermatosi lichenoide purpurea pigmentata di Gougerot e Blum | Papule lichenoidi violacee che si fondono a formare placche | Arti inferiori | Band-come l’infiltrato dermico |
| Lichen aureus | Isolati persistenti rosso-placca arancione e lesioni purpuriche | Arti inferiori | Epidermide invariata e bandacome infiltrato dermico con area di Grenz |
| Malattia di Majocchi | Piastre che si estendono perifericamente con punta telangiectatica ai bordi e dissolvenza nell’area centrale | Arti inferiori e tronco | Identico alla malattia di Schamberg |
Mentre alcuni autori1,13 considerano la capillarite una caratteristica distintiva delle DPP, Ackerman40 non crede che sia questo il caso in quanto vi è un’assenza di fibrina nella parete luminale e di trombi nei lumi.
Diagnosi
Oltre alla biopsia cutanea, si raccomanda un esame del sangue per escludere trombocitopenia, disturbi della coagulazione o autoimmuni (anticorpi antinucleo, fattore reumatoide) e infezioni croniche (anti-HCV e anti-HBsAg).2
Diagnosi differenziale
La diagnosi differenziale deve includere altre condizioni con manifestazioni purpuriche che coinvolgono le estremità inferiori. Queste entità e le loro caratteristiche principali sono riassunte nella tabella 3.
Diagnosi differenziale per le dermatosi purpuriche pigmentate.
| Entità cliniche | Caratteristiche principali | |
|---|---|---|
| Reazioni direazioni di ipersensibilità ai farmaci | Uso recente del farmaco causale | |
| Carbamazepina, meprobamato, clordiazepossido, furosemide, nitroglicerina, vitamina B1, e 5-fluorouracile topico | ||
| Dermatite purpurea da contatto agli indumenti | Lesioni limitate alle aree della pelle in contatto con gli indumenti; prurito intenso | |
| Lana, coloranti | ||
| Porpora da stasi venosa | Segni di insufficienza venosa cronica: gonfiore, vene varicose, sensazione di pesantezza, ulcere venose | |
| Deposito di emosiderina nel derma profondo | ||
| Porpora dovuta a trombocitopenia | Associata alla conta piastrinica | |
| Porpora senile | In pazienti anziani, la porpora può essere associata all’uso di antipiastrinici, anticoagulanti o uso di corticosteroidi | |
| Esantema purpurico dovuto a infezione virale | Altri segni di infezione | |
| Vasculite leucocitoclastica | Lesioni purpuriche palpabili | |
| All’istologia: necrosi fibrinoide, rigonfiamento endoteliale e leucocitosi | ||
| Porpora di Schönlein-Henoch | Età, 3-15 anni; porpora simmetrica che colpisce gambe e natiche; dolori articolari e addominali | |
| Sarcoma di Kaposi | Affligge pazienti anziani o immunodepressi | |
| Su istologia: cellule fusate nel derma che formano lumi vascolari irregolari | ||
| Micosi fungoide purpurea | Inizio > 1 anno; disseminata, profilo monoclonale nell’infiltrato e perdita di CD7 |
Fonte: Sardana et al.2, Kim et al.,25 e Risikesan et al.26
Dermoscopia
Il reperto dermoscopico più comune è un diffuso sfondo rosso ramato che istopatologicamente corrisponde all’infiltrato linfocitario dermico, ai globuli rossi extravasati e ai macrofagi carichi di emosiderina.41 Altri risultati includono globuli e punti rossi, che possono essere spiegati dai RBC extravasati, dall’aumento del numero di vasi sanguigni e dalla dilatazione di questi vasi42 (Fig. 7). I punti marroni si osservano in quasi il 50% dei pazienti e corrispondono alla disposizione sferica o ellittica dei melanociti e dei melanofagi alla giunzione dermoepidermica. In un terzo dei casi, la dermoscopia mostra una pseudo-rete pigmentata che corrisponde allo strato iperpigmentato delle cellule basali e all’incontinentia pigmenti nel derma papillare. I risultati dermoscopici specifici riportati per il lichen aureo includono uno sfondo rosso-rame con punti e globuli marroni e rossi, punti grigi e una pseudo-rete che comprende linee pigmentate interconnesse43 (Fig. 8).
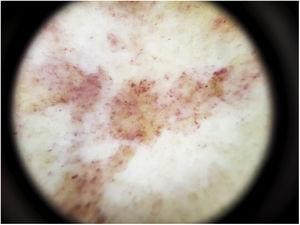
Caratteristiche dermoscopiche della malattia di Schamberg. Sfondo rosso rame e globuli rossi.
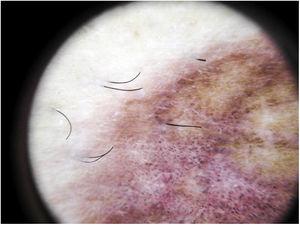
Caratteristiche dermoscopiche del lichen aureo. Sfondo rosso-brunastro con punti grigi e una pseudo-rete pigmentata.
Trattamento
Considerando che le DPP sono benigne e non esistono trattamenti standardizzati di provata efficacia, i rischi e i benefici di qualsiasi trattamento devono essere attentamente soppesati.
Data la natura benigna e largamente asintomatica delle DPP, nessun trattamento è un’opzione.4 Il trattamento, tuttavia, è spesso richiesto a causa della natura cronica della malattia, del suo impatto fisico e psicologico, e della presenza di lesioni estese o prurito.
La maggior parte delle raccomandazioni di trattamento sono basate su piccole serie di casi, e nessuna di esse è supportata da prove sufficienti per essere considerata un trattamento universale.
Una serie di trattamenti topici e sistemici, descritti di seguito, sono stati descritti in piccole serie e case report.
Trattamenti topiciCorticosteroidi topici
I corticosteroidi topici sono i trattamenti più comuni descritti e sono stati osservati per ridurre il prurito e in alcuni casi cancellare le lesioni.2,4
Gli agenti più utilizzati sono i corticosteroidi a media e alta potenza (clobetasolo e metilprednisolone aceponato)
Inibitori topici della calcineurina
L’applicazione topica di tacrolimus44 e pimecrolimus45 per diversi mesi ha risolto il lichen aureus.
Data la natura cronica delle lesioni da PPD e la necessità di un trattamento a lungo termine, gli inibitori della calcineurina possono essere considerati una buona alternativa ai corticosteroidi topici per schiarire o risolvere le lesioni.
Fototerapia
La fototerapia è una buona opzione per il trattamento della malattia estesa o della PPD che non risponde ai corticosteroidi topici o agli inibitori della calcineurina.
È stato postulato che la fototerapia può essere efficace perché produce un effetto immunomodulatore che modifica l’attività delle cellule T e riduce la produzione di interleuchina 2, con conseguente miglioramento.46
Il trattamento con psoralene e UV-A (PUVA) è stato utilizzato con successo in pazienti con malattia di Schamberg, dermatosi lichenoide purpurica e lichen aureo. Nelle serie pubblicate fino ad oggi, tra 7 e 29 sessioni con dosi cumulative che vanno da 16 a 49 J/cm2 sono state necessarie per ottenere la remissione. Anche il ritiro si è dimostrato efficace, e in alcuni casi è stato necessario un trattamento di mantenimento per diversi mesi per prolungare la risposta.2,47-49
La fototerapia UV-B a banda stretta con dosi cumulative comprese tra 11 e 49 J/cm2 somministrate in 24-60 sessioni ha prodotto risposte favorevoli in pazienti con diverse varianti cliniche di DPP. Come con il trattamento PUVA, ci sono state segnalazioni di recidive dopo l’interruzione del trattamento, ma una buona risposta al ritrattamento.46,50,51
La terapia UV-B a banda stretta è considerata una buona opzione a causa dei suoi pochi effetti avversi e del buon profilo di tollerabilità. Dovrebbe quindi essere tenuta in considerazione come opzione per i pazienti pediatrici, pazienti con lesioni estese e pazienti resistenti ai trattamenti topici.5,52
Trattamenti sistemiciPentoxifillina
Ci sono state segnalazioni di PPD che rispondono alla pentossifillina orale. È stato suggerito che la pentossifillina possa essere efficace perché inibisce l’adesione delle cellule T all’endotelio vascolare attraverso l’interazione con ICAM-1.53,54
La pentossifillina è stata usata da sola, alla dose di 400 mg due o tre volte al giorno per 2 o 3 mesi,42,43 o in combinazione con altri farmaci come le prostacicline (prostaglandina I1)55 e i corticosteroidi orali.56 Anche la pentossifillina è risultata inefficace nel trattamento della DPP.57
Acido ascorbico e bioflavonoidi (Rutina/Rutoside)
Poiché l’acido ascorbico e i bioflavonoidi aumentano la produzione di collagene, riducendo così la permeabilità vascolare e migliorando la funzione della barriera vascolare endoteliale, alte dosi di vitamina C combinate con un flavonoide glicoside (come la rutoside/rutina), presente negli agrumi, somministrate per diversi mesi hanno portato a miglioramenti clinici e in alcuni casi alla risoluzione.58
Altri trattamenti
Ci sono stati rapporti isolati di risposta a vari trattamenti sistemici, come la griseofulvina,59 la colchicina,60 il metotrexato,61 e la ciclosporina.62
Conclusioni
Le DPP sono una condizione dermatologica comune e hanno un grande impatto sulla qualità della vita del paziente a causa sia dei sintomi che delle preoccupazioni cosmetiche. Sebbene le diverse varianti siano clinicamente molto simili, esistono una serie di caratteristiche cliniche, istopatologiche e dermoscopiche che aiutano a stabilire una diagnosi più specifica.
Infine, sebbene non ci siano prove sufficienti in letteratura per raccomandare un qualsiasi trattamento come prima linea, esistono numerose opzioni che possono ottenere notevoli miglioramenti.
Conflitti di interesse
Gli autori dichiarano di non avere conflitti di interesse.