R. Kishore Kumar y Surinder Paul Negi*
Del Departamento de Pediatría y *Medicina de Urgencias, Hospital Regional del Noroeste y Universidad de Tasmania, Australia.
Solicitudes de reimpresión: Dr. R. Kishore Kumar, pediatra consultor y profesor de pediatría y salud infantil, North West Regional Hospital and University of Tasmania, P O Box 258, Burnie TAS 7320, Australia. E-mail: [email protected]
ManuscritoRecibido: 5 de abril de 1999;
Revisión inicial completada: 26 de mayo de 1999;
RevisiónAceptada: 18 de agosto de 1999
El síndrome de triploidía es un síndrome poco frecuente y se estima que se produce en aproximadamente el 2% de los conceptus(1). La identificación del síndrome es de suma importancia ya que la mayoría de ellos mueren dentro de los primeros 2 meses, y también para la familia para el posterior asesoramiento genético, la planificación de la futura familia y para su duelo. Informamos de un caso para resaltar estas cuestiones.
Informe del caso
Un recién nacido fue trasladado a nuestro hospital desde un centro periférico para una evaluación adicional debido a la «apariencia inusual». El bebé era el segundo hijo de una pareja caucásica no consanguínea. La madre tenía 30 años. Tienen un niño de 3 años, que está sano y bien. El segundo embarazo había abortado espontáneamente a las 14 semanas de gestación. No había antecedentes familiares de enfermedades importantes ni de otros trastornos. El bebé era pequeño para la edad gestacional, con una grave deficiencia de crecimiento intrauterino y un peso al nacer de 1.600 g a las 41 semanas de gestación. Tenía una fontanela anterior grande y una fontanela posterior, orejas de implantación baja, hipertelorismo, nariz puntiaguda y paladar alto. Había sindactilia del tercer y cuarto dígitos tanto en las extremidades superiores como en las inferiores. Se observó un pliegue simiesco en ambas manos. Los testículos no habían descendido en ambos lados y la longitud del pene era inferior a 0,5 cm. Sólo se observaban dos vasos umbilicales. También tenía los pies en forma de balancín en ambos lados. También tenía una pequeña comunicación interventricular confirmada en un ecocardiograma bidimensional. En vista de todo lo anterior, se sospechó que tenía una trisomía 18. La ecografía renal era normal. Su estudio cromosómico reveló una deleción 18p, pero por lo demás no había evidencia de ninguna trisomía. En todas las células analizadas, un cromosoma 18 homólogo aparecía atípico dentro de la región centrométrica del brazo corto. En vista de ello, se realizó un estudio cromosómico parental que reveló cromosomas maternos con deleción 18p, exactamente igual que el bebé. Sin embargo, la madre es fenotípicamente normal, sin evidencias de dismorfismo y sin discapacidad mental. Por lo tanto, los genetistas lo atribuyeron a un hallazgo incidental. El bebé fue revisado posteriormente por nuestro genetista clínico senior, que diagnosticó el síndrome de triploidía y se realizó una biopsia de piel. El cultivo de fibroblastos de la piel reveló un cariotipo mosaico 46XY/69XXY, consistente con el diagnóstico de síndrome de triploidía mosaico.
Dado que nuestro caso es una triploidía mosaico, el bebé sigue vivo a la edad de 7 meses pesando sólo 3,17 kg con un marcado retraso en el desarrollo. Los padres han aceptado la realidad y la han asumido. Desde el diagnóstico, el número de visitas a urgencias por «problemas de alimentación y falta de peso» ha sido nulo, en comparación con las 10 visitas realizadas en las primeras 8 semanas al médico de cabecera. La familia, consciente de que el riesgo de recurrencia es insignificante, espera su tercer hijo con muy poca aprensión, aunque tiene previsto realizar una amiocentesis a las 16 semanas de gestación para confirmarlo. El diagnóstico de este bebé con un síndrome de triploidía, hizo que la
familia aceptara y tuviera tiempo suficiente para hacer el duelo, planificar su próximo hijo de una manera mucho más relajada; ayudó al médico de cabecera a aconsejar a la familia de forma adecuada y al pediatra a organizar un asesoramiento apropiado para su próximo hijo.
Discusión
El síndrome de triploidía se caracteriza por una dismadurez general, hipotonía muscular, fontanela posterior grande, pabellones auriculares dismórficos de implantación baja, hipertelorismo, microftalmia y colobomata, sindactilia cutánea del tercer y cuarto dedo, pliegue simiesco, hipospadias y/o genitales externos mal desarrollados(2). La mayoría de los bebés nacen prematuramente y suelen vivir menos de un día. Puede haber evidencia de una placenta grande con frecuentes cambios hidatiformes. Todos los casos de triploidía completa han nacido muertos o han muerto en el período neonatal temprano(3), siendo cinco meses la supervivencia más larga registrada(4).
No se puede exagerar la importancia de un diagnóstico confirmado correcto para cualquier niño, en particular para una familia que se enfrenta a un bebé «dismórfico» y que está planeando tener más hijos. Los pediatras reconocen fácilmente los trastornos sindrómicos que implican trisomías de cromosomas. La cuestión surge cuando los cromosomas son normales, cuando la gente asume que se trata de un problema «idiopático». El síndrome de triploidía es uno de ellos, en el que las líneas celulares triploides pueden haber desaparecido de la sangre periférica y la evidencia de la triploidía sólo puede encontrarse en los fibroblastos de la piel cultivados. El Síndrome de Palliester Killian es otro de estos síndromes en el que se ha documentado la tetrasomía 12p en los fibroblastos de la piel de los individuos afectados, pero no en la sangre periférica. En la mayoría de los casos de síndrome de triploidía, el conjunto extra de cromosomas es de origen paterno, y el 66% se atribuye a la doble fecundación, el 24% a la fecundación con un espermatozoide diploide y el
10% a la fecundación de un óvulo diploide. La edad materna avanzada no ha sido un factor a diferencia de la mayoría de las trisomías. No hay datos que indiquen un mayor riesgo, como el que se observa con la no disyunción cromosómica, lo que significa que se puede ser más optimista a la hora de ofrecer asesoramiento genético para los futuros hijos. Pero en varios casos, un embarazo triploide es seguido o precedido por un embarazo molar.
Hay que pensar en esta posibilidad más bien pronto que tarde, en vista de la reducida vida de estos bebés, para poder hacer las pruebas apropiadas y ofrecer asesoramiento genético a la familia implicada para darles suficiente tiempo para el duelo y para que asuman la realidad y también para planificar futuros embarazos sin ser aprensivos. Una búsqueda bibliográfica ha revelado hasta ahora sólo 17 citas sobre este tema en el mundo, siendo la supervivencia más larga de 5 meses. Comunicamos este caso para resaltar su existencia y la prolongada supervivencia de nuestro caso probablemente debido al mosaicismo.
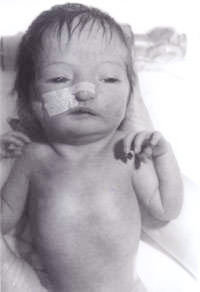
Fig. 1. Bebé con síndrome de triploidía.
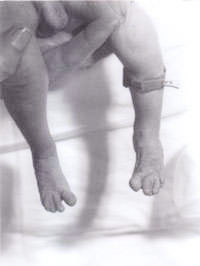
Fig. 2. Sindactalía del tercer y cuarto dedo del pie.
Agradecimientos
Los autores desean agradecer a la Dra. Agnes Bankier, Genetista Clínica Senior y al Dr. Stephen Robertson, Becario de Genética Clínica en el Royal Children’s Hospital de Melbourne por su ayuda en el diagnóstico y asesoramiento genético de la familia.
1. Jones KL. Síndrome de triploidía y síndrome de mixoploidía diploide/triploide. En: Smith’s Recognizable Patterns of Human Malformation, 4th edn. Philadelphia, 1988; pp 32-33.
2. Leisti JT, Raivio KO, Rapola MH, Saksela EJ, Aula PP. El fenotipo de la triploidía humana. Birth Defects Orig Artic Ser 1974; 10: 248-253.
3. Garzena E, Farinasso D, Prandi GM, Vardeu P, Bagna R, Cavo L, et al. Triploidy syndrome: A propósito de un caso. Minerva Pediatr 1995; 47: 307-311.
4. Arvidsson CG. Un niño con tripolidia completa y una supervivencia inusualmente larga. Acta Pediatr Scand 1986; 75: 507-510.