La neuropatia motoria multifocale (MMN) è una rara neuropatia motoria con un range di prevalenza riportato di 0.3-3 casi su 100.000.1 Colpisce più i maschi che le femmine (2,7:1) e l’esordio avviene solitamente prima dei 50 anni di età.1 La malattia può progredire fino alla debolezza permanente e alla disabilità, ma non è pericolosa per la vita o invalidante come la sclerosi laterale amiotrofica (SLA, una delle malattie dei motoneuroni).2 I sintomi della MMN si sovrappongono ad altre malattie a predominanza motoria, come la poliradicoloneuropatia demielinizzante infiammatoria cronica (CIDP), la SLA e le varianti della SLA (atrofia muscolare progressiva, sindromi del braccio e della gamba fluttuanti); questa sovrapposizione può introdurre incertezza diagnostica.
Riconoscere la significativa sovrapposizione nella presentazione della MMN e della SLA è particolarmente importante dato che la MMN è curabile, mentre la SLA è rapidamente mortale e non trattabile. Il profondo impatto psicosociale di una diagnosi di SLA è ben noto e comprende dolore, depressione, ansia, sentimenti di disperazione e altri effetti psicologici negativi che alterano la vita.3,4 Mentre la MMN può essere debilitante, non porta la stessa prognosi grave della SLA e offre la speranza di opzioni di trattamento.
La SLA progredisce inesorabilmente, diffondendosi per coinvolgere più funzioni motorie diverse prima di portare infine alla morte. Il tempo mediano di sopravvivenza è di 3-5 anni e solo il 10% circa sopravvive fino a 10 anni.5,6 Le uniche opzioni di trattamento che modificano la malattia per la SLA sono il riluzolo e l’edaravone.7-12 Al contrario, la MMN è associata a un’aspettativa di vita normale e ha diverse opzioni di trattamento disponibili,13-15 sebbene la MMN possa causare una progressiva debolezza muscolare che può portare a una grave disabilità se non trattata.16 Il trattamento precoce con immunoglobuline intravenose (IVIg) è essenziale per garantire una risposta ottimale al trattamento e prevenire la progressione verso la perdita assonale.17 È quindi fondamentale che la MMN sia diagnosticata correttamente il più presto possibile, consentendo l’inizio di una terapia adeguata per prevenire effetti permanenti, ridurre la disabilità ed evitare il disagio psicologico di una diagnosi errata di SLA, una malattia uniformemente fatale. Sfortunatamente, la MMN può essere molto difficile da diagnosticare in certi casi, in particolare all’inizio del decorso della malattia e in assenza di un evidente blocco di conduzione (CB) e di anticorpi GM1.
Questo articolo mira a discutere la diagnosi differenziale di MMN e SLA, attraverso una serie di casi di studio illustrativi.
Caratteristiche cliniche della neuropatia motoria multifocale e della sclerosi laterale amiotrofica
La SLA ha un esordio insidioso e colpisce più uomini che donne.1 La maggior parte delle persone che sviluppano la malattia ha un’età compresa tra i 40 e i 70 anni, con un’età media di 55 anni al momento della diagnosi.18 Tuttavia, la SLA si manifesta anche in persone di vent’anni e trent’anni.1 L’esordio è asimmetrico, con debolezza che si sviluppa in una regione focale del viso, del braccio o della gamba. I pazienti mostrano segni di spasticità, rapida atrofia muscolare, indebolimento e deperimento. Questo porta all’incapacità di camminare o di muovere le braccia. L’indebolimento muscolare progredisce fino ai muscoli del torace, portando infine all’insufficienza respiratoria. Come la MMN, la SLA può presentarsi con una caduta del piede o debolezza e atrofia dell’estremità superiore distale, anche se è più probabile che coinvolga una porzione di un arto piuttosto che una singola distribuzione nervosa. Come nella SLA, i pazienti con MMN possono manifestare atrofia muscolare e fascicolazioni, anche se le fascicolazioni sono più evidenti nella SLA (Tabella 1).1,19
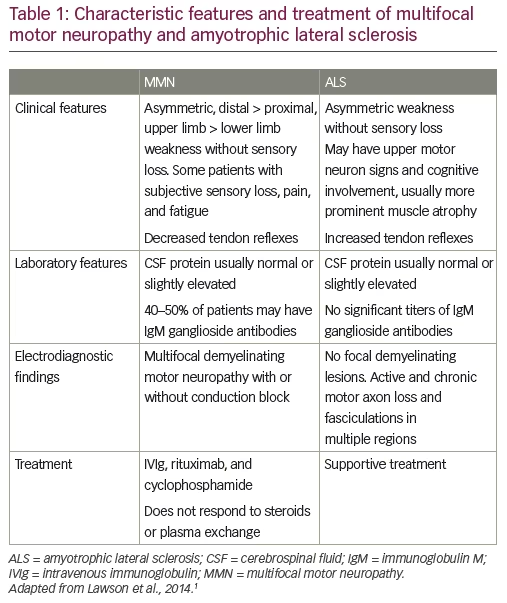
Mentre la diagnosi precoce di MMN può essere difficile nei casi atipici, ci sono caratteristiche cliniche fondamentali che possono aiutare a stabilire la diagnosi. La MMN è caratterizzata da una debolezza asimmetrica degli arti senza perdita sensoriale, che colpisce più comunemente le estremità superiori. La debolezza è frammentaria e multifocale, corrispondente alla distribuzione di singoli nervi piuttosto che segmentale o radicolare (Tabella 1).1,20,21 Come la SLA, la malattia ha un decorso progressivo, ma la progressione della debolezza tende ad essere graduale, piuttosto che insidiosa. I criteri clinici fondamentali richiedono il coinvolgimento di almeno due nervi motori separati senza anomalie sensoriali oggettive, ad eccezione di una lieve compromissione del senso vibratorio. Un modello elettrofisiologico caratteristico è il rallentamento focale e il CB delle fibre nervose motorie all’interno dei segmenti nervosi. La stimolazione della radice del nervo cervicale è anche una tecnica importante per valutare i CB, dato che circa il 13% di tutti i CB nella MMN sono prossimali e possono mancare con gli studi di conduzione nervosa di routine.22,23
A differenza della SLA, che si ritiene sia causata da una combinazione di fattori genetici e ambientali, la MMN ha chiaramente un’eziologia autoimmune; è associata a livelli elevati di IgM anti-GM1 in circa il 50% dei pazienti e risponde al trattamento immunomodulatorio.21,24-26 La fisiopatologia della MMN e della CB è stata trattata altrove e non sarà rivista in questa sede.24
Diagnosi della neuropatia motoria multifocale e della sclerosi laterale amiotrofica
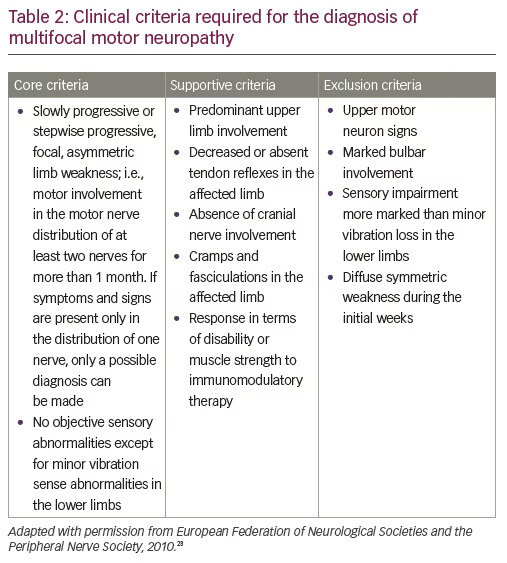
Una diagnosi definitiva di MMN si basa sui criteri clinici di base stabiliti dalla European Federation of Neurological Societies (EFNS)/Peripheral Nerve Society (PNS) (i criteri fondamentali, di supporto e di esclusione nella diagnosi di MMN sono definiti dalla EFNS; Tabella 2).28 I criteri fondamentali comprendono una debolezza degli arti lentamente progressiva o progressiva a tappe, focale, asimmetrica, nella distribuzione di almeno due nervi, per più di un mese; e nessuna anomalia sensoriale oggettiva, eccetto anomalie vibratorie minori negli arti inferiori.
Le caratteristiche importanti che escluderebbero una diagnosi di MMN includono la perdita sensoriale o sintomi sensoriali diversi da una lieve perdita vibratoria nelle dita dei piedi, o una leggera parestesia; così come la debolezza simmetrica all’inizio.29 Tuttavia, anche questi sarebbero atipici per la SLA. In termini di differenziazione dalla SLA e da altri disturbi neuropatici a predominanza motoria, altri criteri di esclusione per la MMN che aiuterebbero a distinguerla dalla SLA includono segni del motoneurone superiore e debolezza bulbare. Gli anticorpi IgM anti-ganglioside GM1 sono presenti in circa la metà dei pazienti con MMN (30-80% a seconda della serie).30,31 Tuttavia, sono stati associati anche ad altre neuropatie immunomediate, neuropatie non immunocorrelate e persino a pazienti con SLA.32
Pertanto, gli anticorpi anti-GM1 sono utili dal punto di vista diagnostico, ma non si può fare affidamento su di essi in modo assoluto.
Anche il reperto caratteristico della CB motoria è stato lasciato fuori dai criteri diagnostici EFNS, poiché la CB in pazienti con MMN altrimenti tipica può non essere rilevabile utilizzando i test elettrofisiologici clinici standard, un test che richiede una notevole competenza elettrofisiologica. La CB è definita come il fallimento della propagazione del potenziale d’azione in un determinato sito lungo un singolo assone e la sua presenza al di fuori dei siti abituali di compressione del nervo sui test di conduzione nervosa costituisce il segno distintivo della MMN. I criteri EFNS permettono di definire la CB definitiva e probabile.28 La CB definitiva è definita come segue: “Riduzione negativa dell’area del potenziale d’azione muscolare composto (CMAP) di picco sulla stimolazione prossimale rispetto a quella distale di almeno il 50% qualunque sia la lunghezza del segmento nervoso (mediano, ulnare e peroneo). L’ampiezza del picco CMAP negativo sulla stimolazione della parte distale del segmento con CB motorio deve essere >20% del limite inferiore della norma e >1 mV e l’aumento della durata del picco CMAP negativo da prossimale a distale deve essere ≤30%”. Il CB probabile è definito da: “riduzione dell’area di picco CMAP negativa di almeno il 30% su un lungo segmento (ad es, dal polso al gomito o dal gomito all’ascella) di un nervo dell’arto superiore con aumento della durata del picco CMAP negativo da prossimale a distale ≤30%” o “riduzione dell’area di picco CMAP negativa di almeno il 50% (uguale a quella definita) con un aumento della durata del picco CMAP negativo da prossimale a distale >30%”.28 A differenza della perdita assonale, la CB non sarà rilevabile se localizzata in un sito che non è accessibile dal test di conduzione nervosa tradizionale. La stimolazione della radice prossimale dovrebbe essere considerata in questi casi.22,23 In uno studio su pazienti con pura malattia del motoneurone inferiore senza CB motoria, il 10% ha risposto alla terapia con IVIg.35 Pertanto, l’assenza di CB o di anticorpi GM1 si oppone alla risposta di IVIg, ma non la esclude.
Oltre al CB e ad altre anomalie demielinizzanti, ci possono essere anche prove di perdita degli assoni motori, come una diminuzione del CMAP evocato distalmente e segni di denervazione e reinnervazione all’elettromiografia ad ago.36 La neurografia a risonanza magnetica è un’altra tecnica diagnostica utile; l’allargamento focale e l’aumento dell’intensità del segnale del plesso brachiale sono visibili sulle immagini pesate in T2 nella MMN.37 Inoltre, molti centri stanno utilizzando gli ultrasuoni per rilevare l’allargamento dei nervi; in uno studio recente, l’ecografia ad alta risoluzione dei nervi periferici ha rivelato modelli distinti di allargamento multifocale dei nervi, che possono sostenere una diagnosi di MMN. I risultati dell’ecografia non hanno correlato bene con la gravità clinica o i risultati elettrofisiologici alla presentazione iniziale, ma i cambiamenti nell’Ultrasound Pattern Sum Score (UPSS) hanno correlato bene con il decorso clinico in termini di forza muscolare, come misurato dal Medical Research Council sum score.38 L’ecografia è stata utilizzata per differenziare la MMN da altre neuropatie39 e può anche essere uno strumento utile per il monitoraggio terapeutico.
La diagnosi della SLA utilizza i criteri di El Escorial e l’algoritmo di Awaji.40 Secondo questi criteri, la diagnosi di SLA richiede segni di degenerazione del motoneurone inferiore tramite esame clinico, elettrofisiologico o neuropatologico, e segni di degenerazione del motoneurone superiore tramite esame clinico. Inoltre, ci deve essere la prova della progressiva diffusione dei segni all’interno di una regione o in altre regioni, l’assenza di prove elettrofisiologiche di altri processi patologici e l’assenza di prove di neuroimmagini di altri processi patologici che potrebbero spiegare i segni clinici ed elettrofisiologici osservati.
La significativa sovrapposizione clinica tra questi due processi motori significa che l’errore diagnostico è comune e può essere difficile da evitare. È importante identificare le caratteristiche che aiutano a distinguere la MMN dalla SLA. Alcune di queste differenze sono state menzionate in precedenza, ma meritano ulteriore enfasi. Mentre nella SLA può essere colpita qualsiasi parte del corpo, la MMN si presenta quasi sempre con la caduta del polso o, meno frequentemente, del piede.17 Come tale, la MMN si presenta con una distribuzione “a chiazze”, mentre la SLA coinvolge un segmento come un arto e si diffonde insidiosamente piuttosto che in modo graduale. Il coinvolgimento bulbare e respiratorio si vedono raramente nella MMN ma sono spesso presenti nella SLA. La debolezza muscolare associata alla MMN comporta una minore atrofia, tranne nei casi gravi o in individui che hanno la malattia da molti anni.41 Le fascicolazioni sono presenti sia nella MMN che nella SLA, ma sono più prominenti e diffuse nella SLA. Inoltre, nella SLA, le fascicolazioni non sono necessariamente limitate ai muscoli deboli.1,19 L’assenza di segni del motoneurone superiore è probabilmente uno degli indicatori clinici più importanti della MMN rispetto alla SLA. Tuttavia, anche questo dato deve essere interpretato con cautela, poiché esistono varianti di SLA del motoneurone inferiore che non sono rare e mancano anche di segni del motoneurone superiore (per esempio, atrofia muscolare progressiva). Elettrofisiologicamente, la MMN si distingue dalla SLA per il CB e, per estensione, per il ridotto reclutamento di unità motorie in assenza di lesioni assonali. Il reclutamento ridotto si vede anche nella SLA, ma è generalmente accompagnato da una denervazione più prominente all’elettromiografia ad ago.1,42
La diagnosi di MMN o SLA può richiedere più di un anno, e un ritardo diagnostico è associato a una prognosi peggiore. In uno studio statunitense su 46 pazienti con MMN inviati a un centro neuromuscolare terziario, solo a 6 è stata data in precedenza la diagnosi corretta.2 Il rapporto tra MMN e SLA è di circa 1 a 20, e i pazienti con MMN sono spesso diagnosticati come affetti da SLA.41,43 La corretta diagnosi di MMN spesso richiede il coinvolgimento di uno specialista neuromuscolare con sufficiente esperienza.
Trattamenti per la neuropatia motoria multifocale ed evidenze per il loro uso
Le attuali linee guida EFNS/PNS raccomandano l’IVIg come terapia standard, basata sull’evidenza, per la MMN.44 Una buona risposta all’IVIg si osserva fino all’80% dei pazienti con MMN.21,45 Anche la somministrazione sottocutanea di immunoglobuline ha mostrato efficacia nella MMN ed è più conveniente della somministrazione endovenosa.46-48 Ulteriori sviluppi come la somministrazione facilitata dalla ialuronidasi e le formulazioni concentrate possono facilitare la somministrazione sottocutanea.49
Vari altri trattamenti tra cui ciclofosfamide, rituximab, micofenolato mofetile, beta-interferone, ciclosporina, azatioprina e infliximab sono stati tutti utilizzati per trattare il MMN, ma i dati degli studi clinici non sono sufficienti a supportarne l’uso per questa indicazione.50 Lo scambio di plasma o i corticosteroidi sono inefficaci o dannosi nella MMN, e il loro uso dovrebbe essere evitato.51,52 Anche le ciclosporine ad alte dosi hanno mostrato una certa efficacia, ma hanno problemi di tossicità, e i dati a sostegno del loro uso sono limitati.53 Il rituximab ha mostrato una certa efficacia nei pazienti con MMN, ma i dati sono contrastanti e devono essere confermati in un grande studio clinico.54
Le opzioni terapeutiche per la SLA sono più limitate. Attualmente sono approvati due farmaci che ritardano la progressione della SLA: il riluzolo, farmaco antieccitotossico disponibile da oltre 20 anni,8,9 e l’edaravone, un antiossidante, approvato di recente.55 Tuttavia, l’edaravone ha dimostrato efficacia solo in un sottogruppo di pazienti con SLA in fase iniziale che soddisfano criteri specifici (SLA di grado 1 o 2 nella Japan ALS Severity Classification, punteggi di almeno 2 punti su tutti i 12 elementi della Revised ALS Functional Rating Scale, capacità vitale forzata dell’80% o più, SLA definita o probabile secondo i criteri rivisti di El Escorial, e durata della malattia di 2 anni o meno).56
Studi di casi
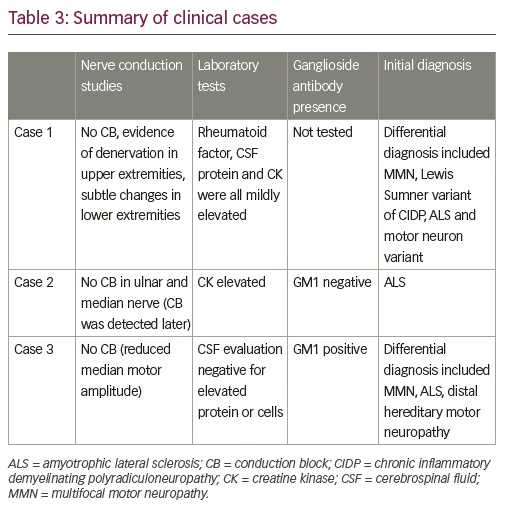
La seguente serie di casi è tratta dall’esperienza degli autori ed è tipica delle sfide nel differenziare la MMN dalla SLA (Tabella 3).
Caso 1
Un uomo di 61 anni si è presentato 7 anni prima con debolezza indolore della mano destra dopo un intervento chirurgico al gomito. Sulla base della presenza di debolezza e atrofia, è stata fatta una diagnosi clinica di neuropatia ulnare.Alla rivalutazione 2 anni dopo, aveva sviluppato una recrudescenza della debolezza della mano destra senza disturbi sensoriali. Anche se senza intorpidimento o parestesia, ha riferito un lieve disagio localizzato al cingolo scapolare nella regione sottoscapolare che si estende ai romboidi e coinvolge il braccio prossimale. Il disagio si placò rapidamente, lasciando il posto a una più profonda atrofia della mano e dell’avambraccio. La lesione del nervo interosseo posteriore è stata sospettata sulla base della debolezza delle dita e dell’estensione del polso, senza debolezza associata all’estensione del gomito o alterazione sensoriale all’esame.
Uno studio elettrodiagnostico eseguito in un ospedale esterno ha suggerito una lesione del nervo radiale e una neuropatia ulnare al gomito; il CB non è stato identificato. Gli altri arti non sono stati studiati. L’impressione ortopedica iniziale era di una neuropatia radiale post-traumatica legata a una lesione al gomito durante l’allenamento di arti marziali. Il paziente è stato rinviato per studi neurologici ed elettrodiagnostici ripetuti. Gli studi di conduzione nervosa hanno rivelato risposte motorie radiali, ulnari e mediane di bassa ampiezza a destra, con conduzione sensoriale conservata, ma non soddisfacevano i criteri per la CB. L’elettromiografia ad ago ha rivelato potenziali di fibrillazione prominenti e onde taglienti positive nei muscoli distali della mano destra; denervazione meno prominente (tr-1+) nei muscoli bicipiti e tricipiti con anomalie di reclutamento neurogenico associate; potenziali di fibrillazione radi e onde taglienti positive nel deltoide e nell’estensore digitorum communis dell’estremità superiore sinistra; e scariche ripetitive complesse nei muscoli gastrocnemio mediale senza prove di denervazione attiva o di cambiamenti cronici delle unità motorie. Un disordine dei motoneuroni o dei loro assoni è stato sospettato sulla base di questi risultati.
La SLA era la diagnosi principale a causa dell’assenza di CB definita; presenza di denervazione attiva in più miotomi dell’estremità superiore destra e meno prominentemente dell’estremità superiore sinistra; e cambiamenti neurogenici più sottili nei muscoli gastrocnemici mediali delle estremità inferiori. Le prove contro la SLA includevano l’assenza di segni del motoneurone superiore, l’assenza di prove di denervazione e reinnervazione cronica, e la mancanza di una progressione significativa nell’arco di 2-3 anni.
I test sierologici accessori hanno rivelato un fattore reumatoide leggermente elevato e la creatinchinasi sierica. Anche le proteine del liquido cerebrospinale (CSF) erano leggermente elevate. La diagnosi differenziale comprendeva la MMN, la variante di Lewis Sumner della CIDP e la SLA o un’altra variante del motoneurone.
È stato iniziato un ciclo di immunoglobuline a intervalli di 3 settimane dopo una dose di carico di 2 g/kg per 5 giorni. Questo ha portato a un miglioramento clinico con un aumento della forza dell’arto superiore destro e un graduale ripristino della massa muscolare nella mano destra.
Questo caso evidenzia alcune importanti difficoltà nel distinguere la MMN dalla malattia del motoneurone. In particolare, l’assenza di CB definitiva può rendere la diagnosi essenzialmente indistinguibile da una variante del motoneurone inferiore della SLA. L’individuazione della CB richiede competenza tecnica, compresa l’attenzione alle misure di distanza, la stimolazione sovramassimale e il posizionamento ottimale degli elettrodi. Anche con la competenza tecnica, CB può non soddisfare i criteri considerati “definiti”, o può essere prossimale ai siti di registrazione. Questo sottolinea l’importanza dell’elettromiografia ad ago come parte della diagnosi elettrofisiologica. In particolare, le anomalie di reclutamento senza attivo (potenziali di fibrillazione e onde taglienti positive) o la denervazione cronica possono essere un indizio importante.
L’assenza di progressione significativa è un ulteriore aspetto importante della diagnosi; la SLA progredisce inevitabilmente, e la diagnosi è confermata se un paziente sviluppa delle caratteristiche che lo definiscono, come la disfunzione bulbare o segni del motoneurone superiore.
Caso 2
Il paziente è un uomo di 70 anni a cui è stata diagnosticata la SLA dopo aver sviluppato una debolezza bilaterale della mano che lo ha reso incapace di lavorare come scultore. Le sue lamentele iniziali riguardavano la debolezza della mano destra, manifestata dalla perdita di destrezza e dalla rapida perdita di massa muscolare nella mano e nell’avambraccio destro. Questo è stato seguito dalla debolezza dell’arto inferiore destro con caduta del piede. Seguì la debolezza dell’estremità inferiore distale sinistra, ma rimase più lieve di quella destra. Il CB non è stato rilevato dallo studio elettrodiagnostico iniziale; solo due nervi motori sono stati studiati (nervo ulnare e mediano a destra). A causa di questi sintomi e dell’assenza di CB sullo studio elettrodiagnostico, gli è stata data una diagnosi di SLA.
Il paziente è stato rinviato per un altro parere quando non è riuscito a progredire come previsto per la sua diagnosi di SLA. Al momento della sua rivalutazione, tre dei suoi quattro arti erano colpiti. La debolezza colpiva soprattutto gli estensori delle dita, gli intrinseci della mano e l’abduzione del pollice a destra. I gruppi muscolari colpiti erano sciupati, ma non c’era perdita sensoriale. Il suo discorso era chiaro senza disartria o disfonia. La lingua era normale senza fascicolazioni. I riflessi di stiramento muscolare erano depressi in tutto. La presenza di crampi muscolari, soprattutto nelle gambe e nel tronco, ha richiesto la risonanza magnetica del midollo toracico, che non ha rivelato alcun miglioramento del midollo spinale o delle radici. Gli studi sierologici hanno rivelato un’elevata creatinchinasi in due occasioni separate. I titoli dei gangliosidi erano negativi per l’anticorpo GM1. La diagnosi di MMN è stata confermata dal ritrovamento di CB in sedi non compressive nel nervo ulnare, radiale e peroneo destro. C’era una certa denervazione attiva nel tibiale anteriore e nel gastrocnemio mediale, ma questa era modesta e non era accompagnata da prove di cambiamenti significativi delle unità motorie. Altri muscoli studiati erano normali con l’eccezione di pronunciate anomalie di reclutamento nei muscoli innervati radiali e ulnari senza evidenza di denervazione attiva.
IVIg è stato avviato e ha portato ad un miglioramento della debolezza in tutte le estremità. La forza dell’arto superiore destro è migliorata ma non è tornata al livello di base. Una valutazione della dipendenza da immunoglobuline (Ig) è stata eseguita ~ 8 mesi dopo l’inizio della regolare IVIg e si era deteriorata per quanto riguarda la forza in entrambi gli arti superiori e inferiori con la ricomparsa della caduta del piede dell’arto inferiore sinistro. L’IVIg regolarmente programmato è stato ripreso e ha portato a un ritorno alla forza osservata prima del test di dipendenza dalle Ig.
Questo caso evidenzia il ruolo della rivalutazione nella valutazione dei pazienti con SLA atipica. La rivalutazione è stata richiesta quando il paziente non ha progredito come previsto per la sua malattia. Le caratteristiche che erano atipiche includevano l’assenza di sintomi bulbari e l’assenza di segni del motoneurone superiore con riflessi diffusamente depressi.
Su una nuova valutazione, il paziente era progredito fino a coinvolgere più nervi motori, e l’evidenza di CB nei nervi motori ulnari e radiali ha confermato la diagnosi. La presenza di mononeuropatie multiple può imitare la distribuzione miotomica della SLA alla presentazione. Questo sottolinea anche l’importanza di mantenere un indice di sospetto all’inizio del corso della malattia per rilevare un modello graduale di debolezza suggestivo di una mononeuropatia multifocale piuttosto che una distribuzione miotomica della debolezza. L’assenza di denervazione sull’elettromiografia ad ago dovrebbe essere un importante indizio diagnostico per una diagnosi alternativa, e le anomalie di reclutamento sproporzionate si adattano alla MMN. Va notato che nella MMN, la CB prossimale può non essere rilevabile dagli studi di conduzione nervosa ma può essere apprezzata dalle anomalie di reclutamento. Inoltre, questo caso sottolinea l’importanza di un adeguato campionamento precoce dei nervi motori con attenzione all’esecuzione di stimolazioni prossimali.
Caso 3
Un uomo di 52 anni si è presentato alla clinica ortopedica con lamentele di “un dito anulare destro che si bloccava” dopo aver usato delle cesoie per un periodo prolungato. La sua debolezza era progredita nel corso dei 12-18 mesi precedenti. Nel rivedere la sua storia, ha ammesso che la progressione era “intermittente”, riportando un brusco peggioramento della forza della mano nei 4-6 mesi precedenti la presentazione. Aveva difficoltà con la flessione del 4° e 5° dito della mano destra e teneva il 5° dito in posizione abdotta. Non poteva tenere il 5° dito in piena estensione e aveva debolezza nell’abduzione delle dita. Non c’era dolore associato, intorpidimento o formicolio, ma si lamentava di crampi localizzati alle braccia, alla parte superiore della schiena e alle spalle.
Era stato indirizzato per una consultazione neurologica e una valutazione elettrodiagnostica a causa dell’atrofia ipotenarica; una diagnosi putativa di neuropatia ulnare è stata suggerita. L’esame neurologico ha rivelato nervi cranici normali senza evidenza di disfunzione bulbare, atrofia della lingua o fascicolazioni. All’esame motorio, aveva una lieve atrofia dei muscoli del braccio destro, dell’ipotenario e dell’avambraccio. Aveva una lieve atrofia dell’omero sinistro. Il test di forza ha rivelato 4-/5 di abduzione del dito destro e 5-/5 nell’estensione del dito; 4-/5 di abduzione del pollice sinistro; e 4/5 di dorsiflessione della caviglia e dell’alluce destro. L’abduzione della spalla, la flessione del gomito e la forza di estensione del gomito erano normali bilateralmente, così come la forza degli arti inferiori prossimali. I riflessi erano 3+ alla rotula, assenti alla caviglia destra, 2+ alla caviglia sinistra, e diminuiti nelle estremità superiori bilaterali. Lo studio elettrodiagnostico non ha identificato la CB, ma l’ampiezza della risposta motoria mediana destra era ridotta. La denervazione era presente nel primo interosseo dorsale ma non era prominente. Non c’erano alterazioni delle unità motorie come polifasie, unità di grande ampiezza o di lunga durata.
Gli anticorpi antiganglioside hanno rivelato una positività GM1 ad alto titolo. La valutazione del CSF era negativa per proteine o cellule elevate. Sulla base della presenza di anomalie di reclutamento prominenti, positività GM1 e una storia di cambiamento graduale nella funzione della mano, è stato completato un processo di IVIg. Una valutazione neurologica ripetuta ~ 6 mesi dopo la terapia Ig ha rivelato un miglioramento della forza di abduzione del pollice a sinistra e della dorsiflessione della caviglia a destra. Anche l’abduzione del dito destro era migliorata, ma in misura minore. Una diagnosi di MMN è stata sospettata sulla base di questa risposta e lui è rimasto sotto IVIg di richiamo.
Prospettive future
Come illustrato da questi esempi di casi, c’è bisogno di ulteriori tecniche per distinguere la SLA dalla MMN. Si è scoperto che le due condizioni presentano profili distinti di citochine e chemochine nei pazienti. Uno studio del 2015 (n = 56) ha trovato differenze nelle caratteristiche infiammatorie del CSF tra i pazienti con MMN e quelli con SLA; in particolare, i livelli di fattore di crescita dei fibroblasti-2 e di fattore stimolante le colonie di granulociti erano elevati nei pazienti con SLA rispetto a quelli con MMN.57
Inoltre, come discusso in precedenza, dati recenti hanno dimostrato che l’ecografia dei nervi ha un’elevata accuratezza diagnostica nella diagnosi differenziale di SLA e MMN, e potrebbe essere superiore agli studi di conduzione nervosa nella diagnosi di MMN in pazienti ospedalizzati con questa diagnosi differenziale.38,58 Un altro studio recente ha suggerito che l’ecografia delle radici cervicali può essere una tecnica utile per supportare la diagnosi di MMN piuttosto che di SLA, anche in assenza di CB.59 Una diagnosi più rapida e accurata può portare a un maggior numero di pazienti che ricevono IVIg o altri trattamenti per la MMN, anche se il risultato a lungo termine di una diagnosi precoce più diffusa rimane sconosciuto.
Riassunto e osservazioni conclusive
L’errata diagnosi di MMN come SLA è un problema importante con gravi implicazioni cliniche per il paziente a causa delle differenze di prognosi e trattamento. In questa revisione, sono state discusse alcune importanti caratteristiche cliniche che possono aiutare a distinguere i due disturbi. In sintesi, le seguenti caratteristiche dovrebbero allertare il medico su una possibile diagnosi di MMN: (i) coinvolgimento distale dell’arto superiore (anche se il medico dovrebbe essere consapevole che questo è un comune sintomo di presentazione della SLA); (ii) multifocale, progressione graduale nella distribuzione di singoli nervi; (iii) assenza di coinvolgimento bulbare/respiratorio; (iv) anomalie CB/demielinizzanti sullo studio elettrofisiologico; (v) assenza di segni del motoneurone superiore; (vi) fascicolazioni rade e (vii) anticorpi GM1.1,20,21,29
Tuttavia, va ripetuto che le varianti della SLA senza segni del motoneurone superiore possono essere estremamente difficili da distinguere dalle sindromi del motoneurone inferiore immuno-responsive come la MMN. La presenza di anticorpi GM1 o CB dovrebbe sempre sollecitare la considerazione di una prova IVIg; dato che la maggior parte dei pazienti risponderà da 8-12 settimane di dosi di richiamo, questo è appropriato data la gravità di un errore di diagnosi. Allo stesso modo, nei pazienti con sindromi pure del motoneurone inferiore senza anticorpi CB o GM1, una prova di IVIg dovrebbe essere considerata.
È necessario diffondere le informazioni sulla diagnosi clinica di MMN. Le precedenti presentazioni di casi “reali” dimostrano come sia possibile evitare una diagnosi errata. Considerazioni importanti sono la necessità di competenza tecnica nei test elettrodiagnostici, poiché la CB definitiva non è sempre presente, e la necessità di ripetere le valutazioni neurologiche. Se diagnosticata correttamente, la MMN spesso risponde bene all’IVIg. Tuttavia, una diagnosi ritardata compromette l’efficacia del trattamento e può comportare una diminuzione della forza muscolare, una disabilità e una prognosi peggiore.