La neuropathie motrice multifocale (NMM) est une neuropathie motrice rare dont la plage de prévalence signalée est de 0.3-3 cas sur 100 000.1 Elle affecte davantage les hommes que les femmes (2,7:1) et son apparition se produit généralement avant 50 ans.1 La maladie peut évoluer vers une faiblesse et un handicap permanents, mais elle ne met pas la vie en danger et n’est pas aussi invalidante que la sclérose latérale amyotrophique (SLA, l’une des maladies du motoneurone).2 Les symptômes de la MMN se chevauchent avec d’autres maladies à prédominance motrice, telles que la polyradiculoneuropathie inflammatoire démyélinisante chronique (PIDC), la SLA et ses variantes (atrophie musculaire progressive, syndromes du bras fléau/de la jambe fléau) ; ce chevauchement peut entraîner une incertitude diagnostique.
La reconnaissance du chevauchement significatif dans la présentation de la MMN et de la SLA est particulièrement importante étant donné que la MMN est traitable, alors que la SLA est rapidement fatale et non traitable. L’impact psychosocial profond d’un diagnostic de SLA est bien établi et comprend le chagrin, la dépression, l’anxiété, les sentiments de désespoir et d’autres effets psychologiques négatifs qui altèrent la vie.3,4 Bien que la MMN puisse être débilitante, elle ne comporte pas le même pronostic grave que la SLA et offre l’espoir d’options de traitement.
La SLA progresse inexorablement, s’étendant pour impliquer de multiples fonctions motrices différentes avant d’entraîner finalement la mort. La durée médiane de survie est de 3 à 5 ans et seulement environ 10% survivent jusqu’à 10 ans.5,6 Les seules options de traitement modificateur de la maladie pour la SLA sont le riluzole et l’edaravone.7-12 En revanche, la MMN est associée à une espérance de vie normale et dispose de plusieurs options de traitement,13-15 bien que la MMN puisse entraîner une faiblesse musculaire progressive pouvant conduire à un handicap sévère si elle n’est pas traitée16. Un traitement précoce à l’immunoglobuline intraveineuse (IVIg) est essentiel pour garantir une réponse optimale au traitement et prévenir la progression vers la perte axonale.17 Il est donc essentiel que la MMN soit correctement diagnostiquée le plus tôt possible, afin de pouvoir commencer un traitement approprié pour prévenir les effets permanents, réduire l’invalidité et éviter la détresse psychologique liée à un diagnostic erroné de SLA, une maladie uniformément mortelle. Malheureusement, la MMN peut être très difficile à diagnostiquer dans certains cas, en particulier au début de l’évolution de la maladie et en l’absence d’un bloc de conduction (BC) évident et d’anticorps GM1.
Cet article vise à discuter du diagnostic différentiel de la MMN et de la SLA, à travers une série d’études de cas illustratives.
Caractéristiques cliniques de la neuropathie motrice multifocale et de la sclérose latérale amyotrophique
La SLA est d’apparition insidieuse et touche davantage les hommes que les femmes1. La plupart des personnes qui développent la maladie sont âgées de 40 à 70 ans, l’âge moyen étant de 55 ans au moment du diagnostic.18 Cependant, la SLA survient également chez des personnes âgées de 20 à 30 ans.1 Le début est asymétrique, la faiblesse se développant dans une région focale du visage, du bras ou de la jambe. Les patients présentent des signes de spasticité, d’atrophie musculaire rapide, d’affaiblissement et de dépérissement. Il en résulte une incapacité à marcher ou à bouger les bras. L’affaiblissement musculaire progresse vers les muscles de la poitrine, entraînant finalement une insuffisance respiratoire. Comme le MMN, la SLA peut se manifester par un pied tombant ou une faiblesse et une atrophie des extrémités supérieures distales, bien qu’elle soit plus susceptible de toucher une partie d’un membre plutôt qu’une seule distribution nerveuse. Comme dans la SLA, les patients atteints de MMN peuvent manifester une atrophie musculaire et des fasciculations, bien que les fasciculations soient plus importantes dans la SLA (tableau 1).1,19
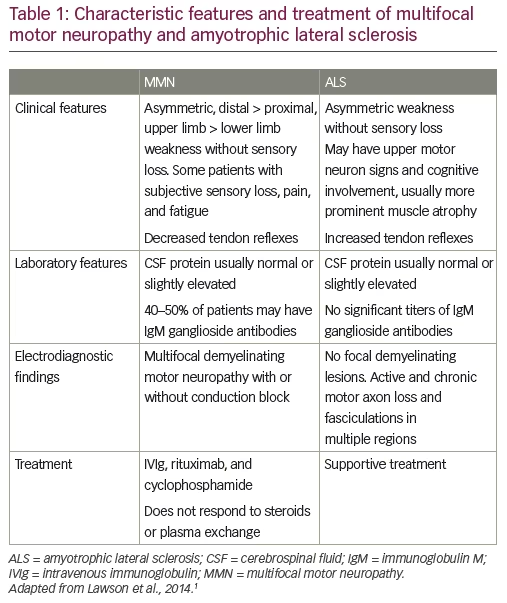
Bien que le diagnostic précoce de MMN puisse être difficile dans les cas atypiques, il existe des caractéristiques cliniques cardinales qui peuvent aider à établir le diagnostic. La MMN se caractérise par une faiblesse asymétrique des membres sans perte sensorielle, affectant plus fréquemment les extrémités supérieures. La faiblesse est inégale et multifocale, correspondant à la distribution de nerfs uniques plutôt que segmentaire ou radiculaire (tableau 1).1,20,21 Comme la SLA, la maladie a une évolution progressive, mais la progression de la faiblesse tend à être graduelle plutôt qu’insidieuse. Les critères cliniques de base exigent l’atteinte d’au moins deux nerfs moteurs distincts sans anomalies sensorielles objectives, à l’exception d’une légère altération du sens vibratoire. Un schéma électrophysiologique caractéristique est le ralentissement focal et le CB des fibres nerveuses motrices dans les segments nerveux. La stimulation de la racine du nerf cervical est également une technique importante pour évaluer le CB, car environ 13 % de tous les CB dans la MMN sont proximaux et peuvent passer inaperçus lors des études de conduction nerveuse de routine22,23.
Contrairement à la SLA, qui serait causée par une combinaison de facteurs génétiques et environnementaux, le MMN a clairement une étiologie auto-immune ; il est associé à des taux élevés d’IgM anti-GM1 chez ~50% des patients et répond à un traitement immunomodulateur.21,24-26 La physiopathologie du MMN et du CB a été couverte ailleurs et ne sera pas revue ici24.
Diagnostic de la neuropathie motrice multifocale et de la sclérose latérale amyotrophique
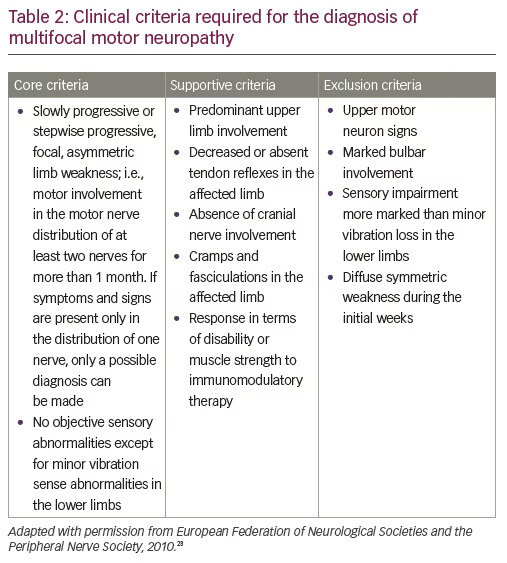
Un diagnostic définitif de MMN est basé sur les critères cliniques de base définis par la Fédération européenne des sociétés de neurologie (EFNS)/Peripheral Nerve Society (PNS) (les critères de base, de soutien et d’exclusion dans le diagnostic de MMN sont définis par l’EFNS ; Tableau 2).28 Les critères fondamentaux comprennent une faiblesse des membres lentement progressive ou progressive par étapes, focale et asymétrique, dans la distribution d’au moins deux nerfs, depuis plus d’un mois ; et aucune anomalie sensorielle objective, à l’exception d’anomalies vibratoires mineures dans les membres inférieurs.
Les caractéristiques importantes qui excluraient un diagnostic de MMN comprennent une perte sensorielle ou des symptômes sensoriels autres qu’une légère perte vibratoire dans les orteils, ou une légère paresthésie ; ainsi qu’une faiblesse symétrique au début29. Cependant, ces éléments seraient également atypiques pour la SLA. En termes de différenciation par rapport à la SLA et à d’autres troubles neuropathiques à prédominance motrice, d’autres critères d’exclusion de la MMN qui aideraient à la distinguer de la SLA sont les signes des motoneurones supérieurs et la faiblesse bulbaire. Les anticorps IgM anti-ganglioside GM1 sont présents chez environ la moitié des patients atteints de MMN (30-80% selon les séries).30,31 Cependant, ils ont également été associés à d’autres neuropathies à médiation immunitaire, à des neuropathies non liées à l’immunité et même à des patients atteints de SLA.32
Par conséquent, les anticorps anti-GM1 sont utiles sur le plan diagnostique, mais ne peuvent pas faire l’objet d’une confiance absolue.
La constatation caractéristique de la CB motrice a également été laissée de côté dans les critères de diagnostic de l’EFNS, car la CB chez les patients présentant une MMN par ailleurs typique peut ne pas être détectable à l’aide de tests électrophysiologiques cliniques standard, un test qui nécessite une expertise électrophysiologique considérable. La NC est définie comme l’échec de la propagation du potentiel d’action à un endroit donné le long d’un seul axone et sa présence en dehors des sites habituels de compression nerveuse lors des tests de conduction nerveuse constitue la marque distinctive de la MMN. Les critères de l’EFNS prévoient un NC certain et probable.28 Le NC certain est défini comme suit : « Réduction négative de la surface du potentiel d’action musculaire composé (CMAP) de pointe lors de la stimulation proximale par rapport à la stimulation distale d’au moins 50 % quelle que soit la longueur du segment nerveux (médian, cubital et péronier). L’amplitude du CMAP de pointe négatif lors de la stimulation de la partie distale du segment avec CB moteur doit être >20% de la limite inférieure de la normale et >1 mV et l’augmentation de la durée du CMAP de pointe négatif proximal par rapport au distal doit être ≤30% ». La CB probable est définie par : » une réduction de la surface du pic négatif de CMAP d’au moins 30 % sur un long segment (par ex, du poignet au coude ou du coude à l’aisselle) d’un nerf d’un membre supérieur avec une augmentation de la durée du pic CMAP négatif proximal à distal ≤30% » ou « Réduction de l’aire du pic CMAP négatif d’au moins 50% (identique à celle d’un cas certain) avec une augmentation de la durée du pic CMAP négatif proximal à distal >30% ».28 Contrairement à la perte axonale, la CB sera indétectable si elle est située dans un site qui n’est pas accessible par les tests de conduction nerveuse conventionnels. La stimulation de la racine proximale doit être envisagée dans ces cas.22,23 Dans une étude portant sur des patients atteints d’une maladie pure du motoneurone inférieur sans CB moteur, 10 % ont répondu au traitement par IgIV.35 Par conséquent, l’absence d’anticorps anti-CB ou anti-GM1 plaide contre la réactivité aux IgIV, mais ne l’exclut pas.
En plus du CB et d’autres anomalies démyélinisantes, il peut également y avoir des preuves de perte de l’axone moteur, telles qu’une diminution de la CMAP évoquée de manière distale et des signes de dénervation et de réinnervation sur l’électromyographie à l’aiguille.36 La neurographie par résonance magnétique est une autre technique diagnostique utile ; un élargissement focal et une augmentation de l’intensité du signal du plexus brachial sont observés sur les images pondérées en T2 dans le cas du MMN.37 En outre, de nombreux centres utilisent l’échographie pour détecter l’élargissement des nerfs ; dans une étude récente, l’échographie à haute résolution des nerfs périphériques a révélé des modèles distincts d’élargissement multifocal des nerfs, ce qui peut appuyer le diagnostic de MMN. Les résultats de l’échographie ne sont pas bien corrélés avec la gravité clinique ou les résultats électrophysiologiques lors de la présentation initiale, mais les changements dans le Ultrasound Pattern Sum Score (UPSS) sont bien corrélés avec l’évolution clinique en termes de force musculaire, telle que mesurée par le Medical Research Council sum score38. L’échographie a été utilisée pour différencier la MMN des autres neuropathies39 et peut également être un outil utile pour le suivi thérapeutique.
Le diagnostic de la SLA emploie les critères d’El Escorial et l’algorithme d’Awaji.40 Selon ces critères, le diagnostic de la SLA nécessite des signes de dégénérescence des motoneurones inférieurs par un examen clinique, électrophysiologique ou neuropathologique, et des signes de dégénérescence des motoneurones supérieurs par un examen clinique. En outre, il doit y avoir des preuves de la propagation progressive des signes au sein d’une région ou vers d’autres régions, l’absence de preuves électrophysiologiques d’autres processus pathologiques et l’absence de preuves neuro-imagerie d’autres processus pathologiques qui pourraient expliquer les signes cliniques et électrophysiologiques observés.
Le chevauchement clinique important entre ces deux processus moteurs signifie que les erreurs de diagnostic sont fréquentes et peuvent être difficiles à éviter. Il est important d’identifier les caractéristiques qui permettent de distinguer le MMN de la SLA. Certaines de ces différences ont été mentionnées précédemment mais méritent d’être soulignées. Alors que n’importe quelle partie du corps peut être affectée dans la SLA, le MMN se présente presque toujours avec une chute du poignet, ou moins fréquemment, une chute du pied.17 Ainsi, le MMN présente une distribution » parcellaire « , alors que la SLA implique un segment tel qu’un membre et se propage insidieusement plutôt que de manière progressive. Les atteintes bulbaires et respiratoires sont rarement observées dans la MMN mais sont souvent présentes dans la SLA. La faiblesse musculaire associée au MMN implique moins d’atrophie, sauf dans les cas graves ou chez les personnes atteintes de la maladie depuis de nombreuses années.41 Les fasciculations sont présentes à la fois dans le MMN et dans la SLA, mais elles sont plus importantes et plus répandues dans la SLA. De plus, dans la SLA, les fasciculations ne sont pas nécessairement limitées aux muscles faibles.1,19 L’absence de signes du motoneurone supérieur est probablement l’un des indicateurs cliniques les plus importants du MMN par rapport à la SLA. Cependant, ce résultat doit également être interprété avec prudence car il existe des variantes de SLA à motoneurones inférieurs qui ne sont pas rares et qui ne présentent pas non plus de signes de motoneurones supérieurs (par exemple, l’atrophie musculaire progressive). Sur le plan électrophysiologique, le MMN se distingue de la SLA par le CB et, par extension, par un recrutement réduit des unités motrices en l’absence de lésion axonale. La réduction du recrutement est également observée dans la SLA, mais elle s’accompagne généralement d’une dénervation plus importante à l’électromyographie à l’aiguille.1,42
Le diagnostic du MMN ou de la SLA peut prendre plus d’un an, et un retard de diagnostic est associé à un plus mauvais pronostic. Dans une étude américaine portant sur 46 patients atteints de MMN adressés à un centre neuromusculaire tertiaire, seuls 6 d’entre eux avaient auparavant reçu le bon diagnostic.2 Le rapport entre MMN et SLA est d’environ 1 à 20, et les patients atteints de MMN sont souvent diagnostiqués comme ayant une SLA.41,43. Le diagnostic correct de la MMN nécessite souvent l’implication d’un spécialiste neuromusculaire disposant d’une expertise suffisante.
Traitements de la neuropathie motrice multifocale et preuves de leur utilisation
Les directives actuelles de l’EFNS/PNS recommandent l’IVIg comme traitement standard, basé sur des preuves, de la MMN.44 Une bonne réponse à l’IVIg est observée chez jusqu’à 80 % des patients atteints de MMN.21,45 L’administration sous-cutanée d’immunoglobulines a également montré son efficacité dans la MMN et est plus pratique que l’administration intraveineuse.46-D’autres développements, tels que l’administration facilitée par la hyaluronidase et les formulations concentrées, pourraient faciliter l’administration sous-cutanée.49
Divers autres traitements, dont le cyclophosphamide, le rituximab, le mycophénolate mofétil, l’interféron bêta, la ciclosporine, l’azathioprine et l’infliximab, ont tous été utilisés pour traiter le MMN, mais les données d’essais cliniques sont insuffisantes pour soutenir leur utilisation dans cette indication.50 Les échanges plasmatiques ou les corticostéroïdes sont inefficaces ou nocifs dans le cas de la NMM, et leur utilisation doit être évitée.51,52 Les ciclosporines à forte dose ont également montré une certaine efficacité mais présentent des problèmes de toxicité, et les données soutenant leur utilisation sont limitées.53 Le rituximab a montré une certaine efficacité chez les patients atteints de NMM, mais les données sont mitigées et doivent être confirmées par un vaste essai clinique.54
Les options de traitement de la SLA sont plus limitées. Actuellement, deux médicaments sont approuvés pour retarder la progression de la SLA : le riluzole, un médicament anti-excitotoxique, qui est disponible depuis plus de 20 ans,8,9 et l’edaravone, un antioxydant récemment approuvé55. Cependant, l’édaravone n’a démontré son efficacité que chez un sous-ensemble de patients atteints de SLA à un stade précoce et répondant à des critères spécifiques (SLA de grade 1 ou 2 dans la classification japonaise de la gravité de la SLA, scores d’au moins 2 points sur les 12 items de l’échelle révisée d’évaluation fonctionnelle de la SLA, capacité vitale forcée de 80 % ou plus, SLA certaine ou probable selon les critères révisés d’El Escorial, et durée de la maladie de 2 ans ou moins).56
Études de cas
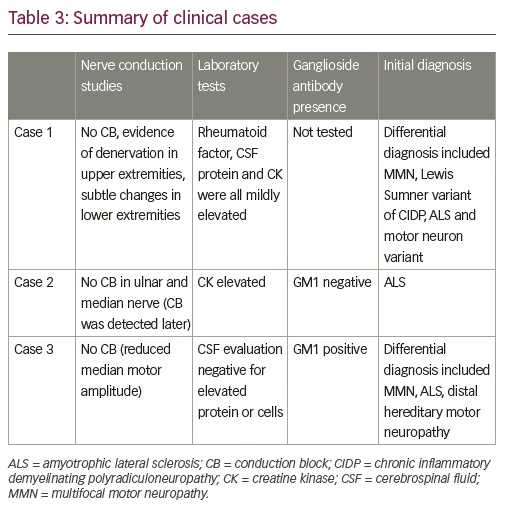
La série de cas suivante est tirée de l’expérience des auteurs et est typique des difficultés à différencier le MMN de la SLA (tableau 3).
Cas 1
Un homme de 61 ans s’est présenté 7 ans auparavant avec une faiblesse indolore de la main droite suite à une chirurgie du coude. Sur la base de la présence d’une faiblesse et d’une atrophie, un diagnostic clinique de neuropathie ulnaire a été posé.Lors de la réévaluation 2 ans plus tard, il avait développé une récurrence de la faiblesse de la main droite sans trouble sensoriel. Bien que sans engourdissement ni paresthésie, il a signalé une légère gêne localisée à la ceinture scapulaire dans la région sous-scapulaire, s’étendant aux rhomboïdes et impliquant le bras proximal. L’inconfort s’est rapidement atténué, laissant place à une atrophie plus profonde de la main et de l’avant-bras. Une lésion du nerf interosseux postérieur a été suspectée sur la base d’une faiblesse d’extension des doigts et du poignet, sans faiblesse d’extension du coude associée ni altération sensorielle à l’examen.
Une étude électrodiagnostique réalisée dans un hôpital extérieur a suggéré une lésion du nerf radial et une neuropathie cubitale au coude ; la CB n’a pas été identifiée. Les autres membres n’ont pas été étudiés. L’impression orthopédique initiale était celle d’une neuropathie radiale post-traumatique liée à une blessure au coude lors d’un entraînement aux arts martiaux. Le patient a été orienté vers un examen neurologique et des études électrodiagnostiques répétées. Les études de conduction nerveuse ont révélé des réponses motrices radiales, cubitales et médianes de faible amplitude à droite, avec une conduction sensorielle préservée, mais ne remplissaient pas les critères de la NC. L’électromyographie à l’aiguille a révélé des potentiels de fibrillation importants et des ondes positives nettes dans les muscles distaux de la main droite ; une dénervation moins importante (tr-1+) dans les muscles biceps et triceps avec des anomalies de recrutement neurogène associées ; des potentiels de fibrillation épars et des ondes positives nettes dans le deltoïde et l’extenseur digitorum communis de l’extrémité supérieure gauche ; et des décharges répétitives complexes dans les muscles gastrocnémiens médians sans preuve de dénervation active ou de modifications chroniques des unités motrices. Un trouble des motoneurones ou de leurs axones a été suspecté sur la base de ces constatations.
La SLA était le diagnostic principal en raison de l’absence de CB définie ; de la présence d’une dénervation active dans plusieurs myotomes de l’extrémité supérieure droite et de façon moins évidente de l’extrémité supérieure gauche ; et de changements neurogènes plus subtils dans les muscles gastrocnémiens médiaux des extrémités inférieures. Les preuves contre la SLA comprenaient l’absence de signes de motoneurones supérieurs, l’absence de preuves de dénervation et de réinnervation chroniques, et l’absence de progression significative sur 2 à 3 ans.
Les tests sérologiques annexes ont révélé un facteur rhumatoïde et une créatine kinase sérique légèrement élevés. Les protéines du liquide céphalo-rachidien (LCR) étaient également légèrement élevées. Le diagnostic différentiel comprenait la MMN, la variante Lewis Sumner de la PIDC et la SLA ou une autre variante du motoneurone.
Un traitement par immunoglobuline a été initié à intervalles de 3 semaines après une dose de charge de 2 g/kg sur 5 jours. Cela a entraîné une amélioration clinique avec une augmentation de la force du membre supérieur droit et une restauration progressive du volume musculaire de la main droite.
Ce cas met en évidence certaines difficultés importantes pour distinguer la MMN de la maladie du motoneurone. En particulier, l’absence de CB définie peut rendre le diagnostic essentiellement indiscernable d’une variante motoneuronale inférieure de la SLA. La détection de la CB exige une compétence technique, notamment une attention particulière aux mesures de distance, une stimulation supramaximale et un placement optimal des électrodes. Même avec une bonne maîtrise technique, il se peut que le CB ne remplisse pas les critères jugés « définitifs », ou qu’il se trouve à proximité des sites d’enregistrement. Cela souligne l’importance de l’électromyographie à l’aiguille dans le cadre du diagnostic électrophysiologique. En particulier, les anomalies de recrutement sans dénervation active (potentiels de fibrillation et ondes aiguës positives) ou chronique peuvent constituer un indice important.
L’absence de progression significative est un autre aspect important du diagnostic ; la SLA progresse inévitablement, et le diagnostic est confirmé si un patient développe des caractéristiques déterminantes telles qu’un dysfonctionnement bulbaire ou des signes du motoneurone supérieur.
Cas 2
Le patient est un homme de 70 ans chez qui on a diagnostiqué la SLA après avoir développé une faiblesse bilatérale des mains qui l’a empêché de travailler comme sculpteur. Il se plaignait initialement d’une faiblesse de la main droite, qui s’est manifestée par une perte de dextérité, puis par une perte rapide du volume musculaire de sa main et de son avant-bras droits. S’ensuivit une faiblesse du membre inférieur droit avec un pied tombant. La faiblesse de l’extrémité inférieure distale gauche a suivi mais est restée plus légère que celle de la droite. La CB n’a pas été détectée lors de l’étude électrodiagnostique initiale ; seuls deux nerfs moteurs ont été étudiés (le nerf cubital et le nerf médian à droite). En raison de ces symptômes et de l’absence de CB sur l’étude électrodiagnostique, il a été donné un diagnostic de SLA.
Le patient a été référé pour un autre avis quand il n’a pas progressé comme prévu pour son diagnostic de SLA. Au moment de sa réévaluation, trois de ses quatre membres étaient affectés. La faiblesse affectait plus particulièrement les extenseurs des doigts, les muscles intrinsèques de la main et l’abduction du pouce droit. Les groupes musculaires affectés étaient atrophiés, mais il n’y avait pas de perte sensorielle. Son discours était clair, sans dysarthrie ni dysphonie. La langue était normale sans fasciculations. Les réflexes d’étirement musculaire étaient déprimés partout. La présence de crampes musculaires, surtout dans les jambes et le tronc, a conduit à une imagerie par résonance magnétique de la moelle thoracique, qui n’a pas révélé d’augmentation de la moelle épinière ou des racines. Les études sérologiques ont révélé une élévation de la créatine kinase à deux occasions distinctes. Les titres de gangliosides étaient négatifs pour l’anticorps GM1. Le diagnostic de MMN a été confirmé par la découverte de CB dans des endroits non compressifs du nerf cubital, radial et péronier droit. Il y avait une certaine dénervation active dans le tibialis antérieur et le gastrocnémien médial, mais elle était modeste et ne s’accompagnait pas de preuves de changements significatifs des unités motrices. Les autres muscles étudiés étaient normaux à l’exception d’anomalies de recrutement prononcées dans les muscles innervés par le radial et le cubital sans preuve de dénervation active.
Les IgIV ont été initiées et ont entraîné une amélioration de la faiblesse dans toutes les extrémités. La force du membre supérieur droit s’est améliorée mais n’est pas revenue au niveau de base. Une évaluation de la dépendance à l’immunoglobuline (Ig) a été réalisée ~8 mois après l’initiation des IVIg réguliers et il s’était détérioré en ce qui concerne la force des extrémités supérieures et inférieures avec une récurrence du pied tombant de l’extrémité inférieure gauche. L’administration régulière d’IVIg a été reprise et a entraîné un retour à la force observée avant le test de dépendance à l’Ig.
Ce cas souligne le rôle de la réévaluation dans l’évaluation des patients atteints de SLA atypique. La réévaluation a été déclenchée lorsque le patient n’a pas progressé comme prévu pour sa maladie. Les caractéristiques qui étaient atypiques comprenaient l’absence de symptômes bulbaires et l’absence de signes des neurones moteurs supérieurs avec des réflexes déprimés de façon diffuse.
Lors de la réévaluation, le patient avait progressé pour impliquer plusieurs nerfs moteurs, et des preuves de CB dans les nerfs moteurs ulnaire et radial ont cintré le diagnostic. La présence de multiples mononeuropathies peut imiter la distribution myotomale de la SLA lors de la présentation. Cela souligne également l’importance de maintenir un indice de suspicion tôt dans l’évolution de la maladie pour détecter un modèle de faiblesse progressive suggérant une mononeuropathie multifocale plutôt qu’une distribution myotomique de la faiblesse. L’absence de dénervation à l’électromyographie à l’aiguille devrait être un indice important pour un autre diagnostic, et les anomalies de recrutement disproportionnées correspondent au MMN. Il faut noter que dans le cas du MMN, la CB proximale peut ne pas être détectable par des études de conduction nerveuse mais peut être appréciée par des anomalies de recrutement. En outre, ce cas souligne l’importance d’un échantillonnage adéquat des nerfs moteurs au début avec une attention à la réalisation de stimulations proximales.
Cas 3
Un homme de 52 ans s’est présenté à la clinique orthopédique en se plaignant que » son annulaire droit se coinçait » après avoir utilisé des cisailles pendant une période prolongée. Sa faiblesse avait progressé au cours des 12 à 18 mois précédents. En examinant ses antécédents, il a admis que la progression était « intermittente », signalant une brusque aggravation de la force de la main dans les 4 à 6 mois précédant la consultation. Il avait des difficultés avec la flexion des 4e et 5e doigts de la main droite et tenait son 5e doigt en position d’abduction. Il ne pouvait pas tenir son 5e doigt en pleine extension et présentait une faiblesse de l’abduction des doigts. Il n’y avait pas de douleur, d’engourdissement ou de picotement associés, mais il se plaignait de crampes localisées aux bras, au haut du dos et aux épaules.
Il a été référé pour une consultation neurologique et une évaluation électrodiagnostique en raison d’une atrophie hypothénar ; un diagnostic putatif de neuropathie ulnaire a été suggéré. L’examen neurologique a révélé des nerfs crâniens normaux sans signe de dysfonctionnement bulbaire, d’atrophie de la langue ou de fasciculations. L’examen moteur a révélé une légère atrophie des muscles du thénar, de l’hypothénar et de l’avant-bras droit. Il présentait une légère atrophie du thénar gauche. Les tests de force ont révélé 4/5 en abduction du doigt droit et 5-/5 en extension du doigt ; 4/5 en abduction du pouce gauche ; et 4/5 en dorsiflexion de la cheville et des orteils droits. La force de l’abduction de l’épaule, de la flexion du coude et de l’extension du coude était normale bilatéralement, tout comme la force des membres inférieurs proximaux. Les réflexes étaient 3+ à la rotule, absents à la cheville droite, 2+ à la cheville gauche, et diminués dans les extrémités supérieures bilatérales. L’étude électrodiagnostique n’a pas identifié de CB mais l’amplitude de la réponse motrice médiane droite était réduite. La dénervation était présente dans le premier interosseux dorsal mais n’était pas proéminente. Il n’y avait pas de modifications des unités motrices telles que la polyphasie, les unités de grande amplitude ou de longue durée.
Les anticorps anti-ganglioside ont révélé une positivité GM1 à un titre élevé. L’évaluation du LCR était négative pour une protéine ou des cellules élevées. Sur la base de la présence d’anomalies de recrutement proéminentes, de la positivité du GM1 et d’une histoire de changement progressif de la fonction de la main, un essai d’IVIg a été réalisé. Une nouvelle évaluation neurologique ~6 mois après le traitement par Ig a révélé une amélioration de la force de l’abduction du pouce à gauche et de la dorsiflexion de la cheville à droite. L’abduction du doigt droit était également améliorée, mais à un degré moindre. Un diagnostic de MMN a été suspecté sur la base de cette réponse et il est resté sous IVIg de rappel.
Perspectives futures
Comme l’illustrent ces exemples de cas, il est nécessaire de disposer de techniques supplémentaires pour distinguer la SLA du MMN. On a constaté que les deux conditions présentent des profils de cytokines et de chimiokines distincts chez les patients. Une étude de 2015 (n = 56) a trouvé des différences dans les caractéristiques inflammatoires du LCR entre les patients atteints de MMN et ceux atteints de SLA ; en particulier, les niveaux du facteur de croissance des fibroblastes-2 et du facteur de stimulation des colonies de granulocytes étaient élevés chez les patients atteints de SLA par rapport à ceux atteints de MMN57.
En outre, comme discuté précédemment, des données récentes ont montré que l’échographie nerveuse a une précision diagnostique élevée dans le diagnostic différentiel de la SLA et du MMN, et pourrait être supérieure aux études de conduction nerveuse dans le diagnostic du MMN chez les patients hospitalisés présentant ce diagnostic différentiel.38,58 Une autre étude récente a suggéré que l’échographie de la racine cervicale peut être une technique utile pour soutenir le diagnostic de MMN plutôt que de SLA, même en l’absence de CB.59 Un diagnostic plus rapide et plus précis peut conduire à un plus grand nombre de patients recevant des IVIg précoces ou d’autres traitements MMN, bien que le résultat à long terme d’un diagnostic précoce plus répandu reste inconnu.
Résumé et conclusions
Le diagnostic erroné de MMN en tant que SLA est un problème important avec des implications cliniques sérieuses pour le patient en raison des différences de pronostic et de traitement. Dans cette revue, certaines caractéristiques cliniques importantes ont été discutées qui peuvent aider à distinguer les deux troubles. En résumé, les caractéristiques suivantes doivent alerter le médecin sur un diagnostic possible de MMN : (i) atteinte distale des membres supérieurs (bien que le médecin doive également savoir qu’il s’agit d’un symptôme courant de la SLA) ; (ii) progression multifocale et progressive dans la distribution de nerfs uniques ; (iii) absence d’atteinte bulbaire/respiratoire ; (iv) anomalies CB/démyélinisantes à l’étude électrophysiologique ; (v) absence de signes des motoneurones supérieurs ; (vi) fasciculations éparses et (vii) anticorps GM1.1,20,21,29
Cependant, il convient de répéter que les variantes de la SLA sans signes du motoneurone supérieur peuvent être excessivement difficiles à distinguer des syndromes du motoneurone inférieur immunoréceptifs tels que le MMN. La présence d’anticorps GM1 ou de CB doit toujours inciter à envisager un essai d’IgIV ; étant donné que la plupart des patients répondent après 8 à 12 semaines de doses de rappel, cela est approprié compte tenu de la gravité d’un mauvais diagnostic. De même, chez les patients présentant un syndrome pur du motoneurone inférieur sans anticorps CB ou GM1, un essai d’IVIg doit être envisagé.
Il est nécessaire de diffuser des informations sur le diagnostic clinique du MMN. Les présentations ci-dessus de cas du » monde réel » démontrent comment les erreurs de diagnostic peuvent être évitées. Les considérations importantes sont la nécessité d’une expertise technique dans les tests électrodiagnostiques, car le CB définitif n’est pas toujours présent, et la nécessité de répéter les évaluations neurologiques. Lorsqu’elle est correctement diagnostiquée, la MMN répond souvent bien aux IgIV. Cependant, un diagnostic tardif nuit à l’efficacité du traitement et peut entraîner une diminution de la force musculaire, un handicap et un pronostic plus sombre.