R. Kishore Kumar et Surinder Paul Negi*
Du département de pédiatrie et de *médecine d’urgence, North West Regional Hospitalet Université de Tasmanie, Australie.
Demandes de réimpression : Dr R. Kishore Kumar, pédiatre consultant et maître de conférences en pédiatrie et santé de l’enfant, North West Regional Hospital et University ofTasmania, P O Box 258, Burnie TAS 7320, Australie. Courriel : [email protected]
ManuscriptReçu : 5 avril 1999;
Révision initiale terminée : 26 mai 1999;
RevisionAcceptée : 18 août 1999
Le syndrome de triploïdie est un syndrome rare et on estime qu’il se produit dans environ 2 % des conceptus(1). L’identification de ce syndrome est de la plus haute importance car la plupart d’entre eux meurent dans les 2 premiers mois, et aussi pour la famille pour le conseil génétique ultérieur, la planification de la future famille et pour leur deuil. Nous rapportons un cas pour mettre en évidence ces questions.
Rapport de cas
Un nouveau-né a été transféré à notre hôpital d’un centre périphérique pour une évaluation plus approfondie en raison de » l’apparence inhabituelle « . Le bébé était le deuxième enfant né d’un couple caucasien non-consanguin. La mère était âgée de 30 ans. Ils ont un garçon de 3 ans, qui est en bonne santé et se porte bien. La deuxième grossesse avait spontanément avorté à 14 semaines de gestation. Il n’y avait pas d’antécédents familiaux de maladies importantes ou d’autres troubles. Le bébé était petit pour son âge gestationnel et présentait un grave déficit de croissance intra-utérin. Il pesait 1600 g à la naissance, à 41 semaines de gestation. Il avait une grande fontanelle antérieure et une fontanelle postérieure, des oreilles décollées, un hypertélorisme, un nez pointu et un palais fortement arqué. Il y avait une syndactalie des troisième et quatrième doigts des membres supérieurs et inférieurs. Un pli simien a été noté sur les deux mains. Les testicules étaient non descendus des deux côtés et la longueur du pénis était inférieure à 0,5 cm. Seuls 2 vaisseaux ombilicaux ont été notés. Il avait également des pieds en forme de bascule des deux côtés. Il avait également une petite communication interventriculaire confirmée par une échocardiographie bidimensionnelle. Compte tenu de tout ce qui précède, on a soupçonné qu’il était atteint de trisomie 18. Son échographie rénale s’est révélée normale. Son étude chromosomique a révélé une délétion 18p, mais par ailleurs aucune évidence d’une quelconque trisomie. Dans toutes les cellules analysées, un homologue du chromosome 18 est apparu atypique dans la région centrométrique du bras court. Devant ce constat, une étude chromosomique parentale a été réalisée révélant des chromosomes maternels avec une délétion 18p, exactement la même que celle du bébé. Mais la mère est phénotypiquement normale, sans signe de dysmorphisme et sans handicap mental. Les généticiens ont donc estimé qu’il s’agissait d’une découverte fortuite. Le bébé a ensuite été examiné par notre généticien clinicien senior, qui a diagnostiqué un syndrome de triploïdie et une biopsie de la peau a été effectuée. La culture des fibroblastes de la peau a révélé un caryotype en mosaïque 46XY/69XXY, ce qui correspond au diagnostic de syndrome de triploïdie en mosaïque.
Puisque notre cas est une triploïdie en mosaïque, le bébé est toujours en vie à l’âge de 7 mois et ne pèse que 3,17 kg avec un retard de développement marqué. Les parents ont accepté cette réalité et s’en accommodent. Depuis le diagnostic, le nombre de visites aux urgences pour « problèmes d’alimentation et absence de prise de poids » a été nul, alors qu’au cours des 8 premières semaines, 10 visites ont été effectuées chez le médecin généraliste. La famille, consciente du fait que le risque de récidive est négligeable, attend son troisième enfant avec très peu d’appréhension, même si elle prévoit de faire une amiocentèse à 16 semaines de gestation pour le confirmer. Le diagnostic de ce bébé avec un syndrome de triploïdie, a permis à la
famille d’accepter et d’avoir suffisamment de temps pour faire son deuil, planifier leur prochain enfant de manière beaucoup plus détendue ; a aidé le médecin généraliste à conseiller la famille de manière appropriée et le pédiatre à organiser un conseil approprié pour leur prochain enfant.
Discussion
Le syndrome de triploïdie se caractérise par une dysmaturité générale, une hypotonie musculaire, une large fontanelle postérieure, des oreillettes dysmorphiques à implantation basse, un hypertélorisme, une microphtalmie et des colobomes, une syndactalie cutanée des troisième et quatrième doigts, un pli simien, un hypospadias et/ou des organes génitaux externes mal développés(2). La plupart des bébés naissent prématurément et vivent généralement moins d’un jour. Il peut y avoir des signes d’un grand placenta avec des changements hydatiformes fréquents. Tous les cas de triploïdie complète ont été soit mort-nés, soit sont décédés au début de la période néonatale(3), cinq mois étant la plus longue survie enregistrée(4).
On ne saurait trop insister sur l’importance d’un diagnostic correct confirmé pour tout enfant, en particulier pour une famille confrontée à un bébé « dysmorphique » et qui envisage d’avoir d’autres enfants. Les troubles syndromiques impliquant des trisomies de chromosomes sont facilement reconnus par les pédiatres. La question se pose lorsque les chromosomes sont déclarés normaux, lorsque les gens supposent qu’il s’agit d’un problème « idiopathique ». Le syndrome de triploïdie est l’un de ces syndromes dans lequel les lignées cellulaires triploïdes peuvent avoir disparu du sang périphérique et la preuve de la triploïdie ne peut être trouvée que dans les fibroblastes cutanés cultivés. Le syndrome de Palliester Killian est un autre de ces syndromes où la tétrasomie 12p a été documentée dans les fibroblastes cutanés des individus affectés, mais pas dans le sang périphérique. Dans la plupart des cas de syndrome de triploïdie, l’ensemble supplémentaire de chromosomes est d’origine paternelle, 66 % étant attribués à une double fécondation, 24 % à la fécondation par un spermatozoïde diploïde et
10 % à la fécondation d’un ovule diploïde. L’âge maternel élevé n’a pas été un facteur contrairement à la plupart des trisomies. Il n’y a pas de données indiquant un risque accru, comme celui observé avec la non-dysjonction chromo-somique, ce qui signifie que l’on peut être plus optimiste tout en offrant un conseil génétique pour les futurs enfants. Mais dans plusieurs cas, une grossesse triploïde est soit suivie, soit précédée d’une grossesse molaire.
Il faut penser à cette possibilité plutôt tôt que tard, compte tenu de la durée de vie réduite de ces bébés, afin de pouvoir faire les tests appropriés pour proposer un conseil génétique à la famille concernée afin de lui laisser suffisamment de temps pour faire son deuil et accepter la réalité, et aussi pour planifier d’autres grossesses sans appréhension. Une recherche documentaire n’a révélé que 17 citations sur ce sujet dans le monde à ce jour, la survie la plus longue étant de 5 mois. Nous rapportons ce cas pour souligner son existence et la survie prolongée de notre cas probablement en raison du mosaïcisme.
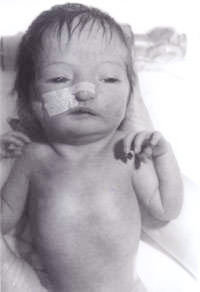
Fig. 1. Bébé présentant un syndrome de triploïdie.
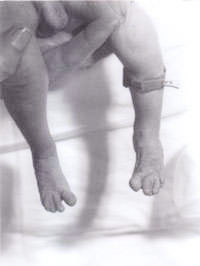
Fig. 2. Syndactalie des troisième et quatrième orteils.
Reconnaissances
Les auteurs tiennent à remercier le Dr Agnes Bankier, généticien clinique principal et le Dr Stephen Robertson, boursier en génétique clinique au Royal Children’s Hospital, Melbourne, pour leur aide au diagnostic et au conseil génétique de la famille.
1. Jones KL. Syndrome de triploïdie et syndrome de mixoploïdie diploïde/triploïde. In : Smith’s Recognizable Patterns of Human Malformation, 4e éd. Philadelphie, 1988 ; pp 32-33.
2. Leisti JT, Raivio KO, Rapola MH, Saksela EJ, Aula PP. Le phénotype de la triploïdie humaine. Birth Defects Orig Artic Ser 1974 ; 10 : 248-253.
3. Garzena E, Farinasso D, Prandi GM, Vardeu P, Bagna R, Cavo L, et al. Triploidy syndrome : A case report. Minerva Pediatr 1995 ; 47 : 307-311.
4. Arvidsson CG. Un garçon avec une triploïdie complète et une survie anormalement longue. Acta Paediatr Scand 1986 ; 75 : 507-510.
.